Error 404 – Законодательство, нормативные акты, образцы документов
Новости:
Все обновления | Последние изменения: 1. «ОралОрал қалалық мәслихатының 2014 жылғы 25 қарашадағы № 30-5 «Орал қаласында аз қамтамасыз етілген отбасыларға (азаматтарға) тұрғын үй көмегін көрсетудің мөлшерін және тәртібін айқындау туралы қағидасын бекіту туралы» шешіміне өзгерістер енгізу туралы Батыс Қазақстан облысы Орал қалалық мәслихатының 2015 жылғы 3 желтоқсандағы № 39-3 шешімі Қазақстан Республикасының 2001 жылғы 23 қаңтардағы «Қазақстан Республикасындағы жергілікті мемлекеттік басқару және өзін-өзі басқару туралы» және 1997 жылғы 16 сәуірдегі «Тұрғын үй қатынастары туралы» Заңдарына сәйк Далее… «ОралОрал қалалық мәслихатының 2014 жылғы 25 қарашадағы № 30-5 «Орал қаласында аз қамтамасыз етілген отбасыларға (азаматтарға) тұрғын үй көмегін көрсетудің мөлшерін және тәртібін айқындау туралы қағидасын бекіту туралы» шешіміне өзгерістер енгізу туралы Батыс Қазақстан облысы Орал қалалық мәслихатының 2015 жылғы 3 желтоқсандағы № 39-3 шешімі Қазақстан Республикасының 2001 жылғы 23 қаңтардағы «Қазақстан Республикасындағы жергілікті мемлекеттік басқару және өзін-өзі басқару туралы» және 1997 жылғы 16 сәуірдегі «Тұрғын үй қатынастары туралы» Заңдарына сәйк Далее…2. ««Қазақстан Республикасы ұлттық қауіпсіздік комитеті органдарының әскери, арнаулы оқу орындарында іске асырылатын жоғары және жоғары оқу орнынан кейінгі білім беру мамандықтары бойынша үлгілік оқу жоспарларын бекіту туралы» Қазақстан Республикасы Ұлттық қауіпсіздік комитеті Төрағасының 2016 жылғы 13 қаңтардағы № 9/ҚБП бұйрығына өзгерістер енгізу туралы» Қазақстан Республикасы Ұлттық қауіпсіздік комитеті Төрағасының 2016 жылғы 10 қазандағы № 67/ҚБП бұйрығы. Қызмет бабында пайдалануға арналған және Деректер базасына енгізілмейді Далее… Қызмет бабында пайдалануға арналған және Деректер базасына енгізілмейді Далее…3. Утверждены Правила согласования размещения предприятий и других сооружений, а также условий производства строительных и других работ на водных объектах, водоохранных зонах и полосах (аннотация к документу от 01.09.2016)Утверждены Правила согласования размещения предприятий и других сооружений, а также условий производства строительных и других работ на водных объектах, водоохранных зонах и полосахАннотация к документу: Приказ Заместителя Премьер-Министра Республики Казахстан – Министра сельского хозяйства Республики Казахстан от 1 сентября 2016 года № 380 «Об утверждении Правил согласования размещения предприятий и других сооружений, а также условий производства строительных и других работ на водных объектах, водоохранных зонах и полосах»В соответствии с подпунктом 7-5) пункта 1 статьи 37 Водного кодекса Республики Казахстан от 9 июля 2003 год Далее…4. Утверждены Правила регулирования цен на услуги, производимые и реализуемые субъектами государственной монополии в области связи (аннотация к документу от 24. 10.2016)Утверждены Правила регулирования цен на услуги, производимые и реализуемые субъектами государственной монополии в области связиАннотация к документу: Приказ Министра информации и коммуникаций Республики Казахстан от 24 октября 2016 года № 221 «Об утверждении Правил регулирования цен на услуги, производимые и реализуемые субъектами государственной монополии в области связи»В соответствии с подпунктом 1) пункта 2 статьи 20 Закона Республики Казахстан от 5 июля 2004 года «О связи» утверждены Далее… 10.2016)Утверждены Правила регулирования цен на услуги, производимые и реализуемые субъектами государственной монополии в области связиАннотация к документу: Приказ Министра информации и коммуникаций Республики Казахстан от 24 октября 2016 года № 221 «Об утверждении Правил регулирования цен на услуги, производимые и реализуемые субъектами государственной монополии в области связи»В соответствии с подпунктом 1) пункта 2 статьи 20 Закона Республики Казахстан от 5 июля 2004 года «О связи» утверждены Далее…5. Утверждены Правила формирования перечня энергопроизводящих организаций, использующих возобновляемые источники энергии (аннотация к документу от 09.11.2016)Утверждены Правила формирования перечня энергопроизводящих организаций, использующих возобновляемые источники энергииАннотация к документу: Приказ Министра энергетики Республики Казахстан от 9 ноября 2016 года № 482 «Об утверждении Правил формирования перечня энергопроизводящих организаций, использующих возобновляемые источники энергии»В соответствии с подпунктом 10-3) статьи 6 Закона Республики Казахстан от 4 июля 2009 года «О поддержке использования возобновляемых источников энергии» утверждены Далее. .. ..6. Изменения внесены в ряд приказов Министра энергетики Республики Казахстан (аннотация к документу от 31.05.2016)Изменения внесены в ряд приказов Министра энергетики Республики КазахстанАннотация к документу: Приказ Министра энергетики Республики Казахстан от 31 мая 2016 года № 228 «О внесении изменений в некоторые приказы Министра энергетики Республики Казахстан»В частности, изменения внесены в приказ Министра энергетики Республики Казахстан «Об утверждении Правил пользования тепловой энергией», изменения затронули понятия и определения используемые в правилах. Также, изменения внесены в ряд пунктов правил, а именно: Далее…7. Заканчивается срок приема заявлений по легализации имуществаЗаканчивается срок приема заявлений по легализации имущества Вниманию всех заинтересованных лиц!Напоминаем, что 31 декабря 2016 года заканчивается легализация имущества, которая проводилась с 1 сентября 2014 года в соответствии с Законом РК от 30 июня 2014 года № 213-V «Об амнистии граждан Республики Казахстан, оралманов и лиц, имеющих вид на жительство в Республике Казахстан, в связи с легализацией ими имущества». При этом, срок подачи документов для легализации недвижимого имущества, находящегося на территории Республики Казахстан, заканчился 30 ноября 2016 года, а для иного имущества срок подачи документов завершается за 5 рабочих дней до конца 2016 года, то есть не позднее 23 декабря 2016 года. Далее… При этом, срок подачи документов для легализации недвижимого имущества, находящегося на территории Республики Казахстан, заканчился 30 ноября 2016 года, а для иного имущества срок подачи документов завершается за 5 рабочих дней до конца 2016 года, то есть не позднее 23 декабря 2016 года. Далее…8. 31 декабря истекает срок уплаты налога на транспорт физическими лицами31 декабря истекает срок уплаты налога на транспорт физическими лицами Вниманию физических лиц, имеющих на праве собственности транспортные средства!Срок уплаты налога на транспортные средства истекает 31 декабря 2016 года.Обратите внимание, что с 1 января 2016 года уплата налога физическими лицами производится по месту жительства.В случае осуществления регистрационных действий по передаче права собственности на транспортное средство, сумма налога, подлежащая уплате за фактический период владения таким объектом лицом, передающим эти права, должна быть внесена в бюджет до совершения указанных действий.Уплата налога на транспортные средства физическим лицом, являю Далее. .. ..9. О дифференциации доходов и расходов населения в Республике Казахстан за 3 квартал 2016 годаО дифференциации доходов и расходов населения в Республике Казахстан за 3 квартал 2016 года По результатам выброчного обследования домашних хозяйств доля населения, имеющего доходы ниже величины прожиточного минимума (уровень бедности), в Республике Казахстан в 3 квартале 2016 года составила 2,5%, по сравнению с соответствующим периодом предыдущего года оставшись на том же уровне. Вместе с тем, по-прежнему, сохраняется разрыв между уровнем бедности среди городского и сельского населения. Наибольшее значение уровня бедности в 3 квартале 2016 года зарегистрировано в Южно-Казахстанской (5,0%), Атырауской и Жамбылско Далее…10. Сагинтаев поручил акимам «удержать» инфляциюСагинтаев поручил акимам «удержать» инфляцию Премьер-министр РК Бакытжан Сагинтаев поручил акимам регионов работать по «удержанию» инфляции в коридоре 6-8%, передает корреспондент Zakon.kz.«В прошлый раз мы говорил о том, что необходимо оставаться в коридоре 6-8% по инфляции. 11 месяц мы грубо так провалили и вот я еще раз обращаюсь к акимам регионов, чтобы в декабре 2016 года мы удержали инфляцию с тем, чтобы остаться в коридоре 6-8%. Работу будем продолжать. На следующей неделе еще поговорим по итогам», – сказал он на заседании Правительства РК.В то же время Глава Кабмина отметил, что тенденция по росту экономики в Казахстане по итогам 11 месяцев положительная.«Мы видим, что хорошие показатели имеем, тенденция положительная. И, если мы по итогам полугодия говорили о том, что было бы хорошо, чтобы мы год Далее… 11 месяц мы грубо так провалили и вот я еще раз обращаюсь к акимам регионов, чтобы в декабре 2016 года мы удержали инфляцию с тем, чтобы остаться в коридоре 6-8%. Работу будем продолжать. На следующей неделе еще поговорим по итогам», – сказал он на заседании Правительства РК.В то же время Глава Кабмина отметил, что тенденция по росту экономики в Казахстане по итогам 11 месяцев положительная.«Мы видим, что хорошие показатели имеем, тенденция положительная. И, если мы по итогам полугодия говорили о том, что было бы хорошо, чтобы мы год Далее…11. Обзор пользователей интернет-услуг ЕНПФ за декабрь 2016 годаОбзор пользователей интернет-услуг ЕНПФ за декабрь 2016 года Количество вкладчиков, выбравших метод веб-информирования Единого накопительного пенсионного фонда, на декабрь 2016 года составляет 2,77 миллиона человек. Доля пользователей онлайн услуг ЕНПФ за год выросла с 13% до 29%.Всего за год число абонентов фиксированного интернета в РК выросло на 201 тысячу, до 2,27 миллиона. Из них 55 тысяч количество новых абонентов сельской местности, всего – 436 тысяч.За 5 лет количество интернет-абонентов в РК выросло почти вдвое – на 93%. При этом показатели села подскочили почти втрое (на 179%). Далее… Из них 55 тысяч количество новых абонентов сельской местности, всего – 436 тысяч.За 5 лет количество интернет-абонентов в РК выросло почти вдвое – на 93%. При этом показатели села подскочили почти втрое (на 179%). Далее…12. Ликвидация организации как основание прекращения производства по гражданскому делу (Тимур Данабаев, практикующий юрист)Ликвидация организации как основание прекращения производства по гражданскому делу Тимур ДанабаевПрактикующий юрист Подпунктом 8) статьи 277 Гражданского процессуального кодекса Республики Казахстан (далее – ГПК РК) предусмотрено, что суд прекращает производство по делу если организация, выступающая стороной по делу, ликвидирована с прекращением ее деятельности и отсутствием правопреемников. Указанные ниже вопросы свидетельствуют о наличии определенных сложностей с толкованием и практическим применением в судебной практике указанной нормы права, а также о существовании различных (нередко противоречивых) подходов к ее применению. Рассмотрим эти Далее. .. ..13. Розничная торговля за ноябрь 2016 годаРозничная торговля за ноябрь 2016 года Средний чек на городского жителя в ноябре 2016 составил 66,2 тысячи тенге – на 7,5% больше, чем годом ранее. Объем ритейла за год вырос на 9,6%, и достиг 669,1 млрд тг.В ноябре объем официальной розничной торговли составил 669,1 млрд тг – на 0,3% (+2,1 млрд тг) больше, чем в октябре, и на 9,6% (+58,7 млрд тг) больше, чем годом ранее.Примечательно, что положительную динамику обеспечили регионы, в то время как обе столицы, концентрирующие 35,5% всего ритейла по РК, в минусе по отношению к октябрю 2016.Наибольший месячный прирост отмечен в Павлодарской области (почти на треть, до 36,5 млрд тг) и Жамбылской области (+17,2%, до 20 млрд тг). Далее…14. Утвержден Генеральный план города Атырау (аннотация к документу от 29.11.2016)Утвержден Генеральный план города Атырау Аннотация к документу: Постановление Правительства Республики Казахстан от 29 ноября 2016 года № 749 «О Генеральном плане города Атырау Атырауской области (включая основные положения)» (не введено в действие)В соответствии со статьей 19 Закона Республики Казахстан от 16 июля 2001 года «Об архитектурной, градостроительной и строительной деятельности в Республике Казахстан» и в целях обеспечения комплексного развития города Атырау Атырауской области Правительство Республики Казахстан утвержден Далее. .. ..15. Реализация кадровой политики в Национальном бюро по противодействию коррупции (аннотация к документу от 21.10.2016)Реализация кадровой политики в Национальном бюро по противодействию коррупцииАннотация к документу: Приказ Председателя Агентства Республики Казахстан по делам государственной службы и противодействию коррупции от 21 октября 2016 года № 18 «О некоторых вопросах реализации кадровой политики в Национальном бюро по противодействию коррупции (Антикоррупционной службе) Агентства Республики Казахстан по делам государственной службы и противодействию коррупции»В соответствии с подпунктом 9) статьи 5-1, Далее…16. Особенности исполнения налогового обязательства при ликвидации и прекращении деятельности (ДГД по Восточно-Казахстанской области, 15 ноября 2016 г.)Особенности исполнения налогового обязательства при ликвидации и прекращении деятельности Законом Республики Казахстан от 29 декабря 2014 года № 269-V «О внесении изменений и дополнений в некоторые законодательные акты Республики Казахстан по вопросам кардинального улучшения условий для предпринимательской деятельности в Республике Казахстан» внесены существенные изменения в части ликвидации предприятий и ИП, а именно, предоставлена возможность закрытия по результатам аудиторской проверки. В Кодекс Республики Казахстан «О налогах и других обязательных платежах в бюджет» (далее- Налоговый кодекс) введена новая статья 37-2 «Ос Далее… В Кодекс Республики Казахстан «О налогах и других обязательных платежах в бюджет» (далее- Налоговый кодекс) введена новая статья 37-2 «Ос Далее…17. Вернуть в административное законодательство (Ержан Карабаев, председатель апелляционной судебной коллеги по уголовным делам Мангистауского областного суда)Вернуть в административное законодательство Ержан Карабаев, председатель апелляционной судебной коллеги по уголовным делам Мангистауского областного суда В судебной практике казахстанских судов возникают проблемные вопросы при рассмотрении уголовных дел по уголовным проступкам и при назначении наказаний за их совершение. Далее…18. Повышая доверие к правосудию (Малик Жаркынбеков, судья Актюбинского областного суда)Повышая доверие к правосудию Малик Жаркынбеков, судья Актюбинского областного суда VII внеочередной Съезд судей Республики Казахстан определил основные направления совершенствования деятельности судов по эффективной защите прав, свобод, достоинства и собственности граждан государства. В целом работа Съезда была нацелена на становление прочной, современной, демократической судебной системы как одной из главных составляющих развития страны, развития нашего государства в среднесрочной и дальней перспективе. Далее… В целом работа Съезда была нацелена на становление прочной, современной, демократической судебной системы как одной из главных составляющих развития страны, развития нашего государства в среднесрочной и дальней перспективе. Далее…19. К эффективной реализации реформ (М. Рысбеков, председатель СМЭС Павлодарской области)К эффективной реализации реформ М. Рысбеков, председатель СМЭС Павлодарской области К 25-й годовщине Независимости мы подходим с новой казахстанской мечтой, которая тождественна главной цели реализуемой нами «Стратегии-2050». К середине ХХІ века мы планируем добиться вхождения Казахстана в число 30 самых развитых государств мира.Лидер нации, выступая на XVI Съезде партии «Нур Отан Далее…20. Снизить размеры взысканий (Ермек Махметов, судья САС г. Актобе)Снизить размеры взысканий Ермек Махметов, судья САС г. Актобе Долгое время, начиная с момента обретения Казахстаном независимости, административному законодательству, регулирующему административно-деликтные правоотношения, не уделялось должного внимания, оно являлось наследием советского времени, сохранив в себе карательно-репрессивный характер. |
СНиП 3.05.05-84 Технологическое оборудование и технологические трубопроводы.
СТРОИТЕЛЬНЫЕ НОРМЫ И ПРАВИЛА
ТЕХНОЛОГИЧЕСКОЕ ОБОРУДОВАНИЕ И ТЕХНОЛОГИЧЕСКИЕ ТРУБОПРОВОДЫ СНиП 3.05.05-84
ГОСУДАРСТВЕННЫЙ КОМИТЕТ СССР ПО ДЕЛАМ СТРОИТЕЛЬСТВА
РАЗРАБОТАНЫ ВНИИмонтажспецстроем Минмонтажспецстроя СССР (инж. В. Я. Эйдельман, д-р техн. наук В. В. Поповский — руководители темы; кандидаты техн. наук В. И. Оботуров, Ю. В. Попов, Р. И. Тавастшерна), Гипронефтеспецмонтажом Минмонтажспецстроя СССР (канд. техн. наук И. С. Гольденберг) и Гипрохиммонтажом Минмонтажспецстроя СССР (инженеры И. П. Петрухин, М. Л. Эльяш).ВНЕСЕНЫ Минмонтажспецстроем СССР.ПОДГОТОВЛЕНЫ К УТВЕРЖДЕНИЮ Отделом технического нормирования и стандартизации Госстроя СССР (инж.
Настоящие правила распространяются на производство и приемку работ по монтажу технологического оборудования и технологических трубопроводов (в дальнейшем — „оборудование” и „трубопроводы”), предназначенных для получения, переработки и транспортирования исходных, промежуточных и конечных продуктов при абсолютном давлении от 0,001 МПа (0,01 кгс/см2) до 100 МПа вкл. (1000 кгс/см2), а также трубопроводов для подачи теплоносителей, смазки и других веществ, необходимых для работы оборудования.Правила должны соблюдаться всеми организациями и предприятиями, участвующими в проектировании и строительстве новых, расширении, реконструкции и техническом перевооружении действующих предприятий.Работы по монтажу оборудования и трубопроводов, подконтрольных Госгортсхнадзору СССР, в том числе сворка и контроль качества сварных соединений, должны производиться согласно правилам и нормам Госгортехнадзора СССР.
(1000 кгс/см2), а также трубопроводов для подачи теплоносителей, смазки и других веществ, необходимых для работы оборудования.Правила должны соблюдаться всеми организациями и предприятиями, участвующими в проектировании и строительстве новых, расширении, реконструкции и техническом перевооружении действующих предприятий.Работы по монтажу оборудования и трубопроводов, подконтрольных Госгортсхнадзору СССР, в том числе сворка и контроль качества сварных соединений, должны производиться согласно правилам и нормам Госгортехнадзора СССР.
1. ОБЩИЕ ПОЛОЖЕНИЯ
1.1. При производстве работ по монтажу оборудования и трубопроводов необходимо соблюдать требования СНиП по организации строительного производства, СНиП III-4-80, стандартов, технических условий и ведомственных нормативных документов, утвержденных в соответствии со СНиП 1.01.01-82*.
1.2. Работы по монтажу оборудования и трубопроводов должны производиться в соответствии с утвержденной проектно-сметной и рабочей документацией, проектом производства работ (ППР) и документацией предприятий-изготовителей.
1.3. Монтаж оборудования и трубопроводов должен осуществляться на основе узлового метода строительства и комплектно-блочного метода монтажа.Примечания: 1. Под узловым методом строительства понимается организация строительно-монтажных работ с разделением пускового комплекса на взаимоувязанные между собой технологические узлы — конструктивно и технологически обособленные части объекта строительства, техническая готовность которых после завершения строительно-монтажных работ позволяет автономно, независимо от готовности объекта в целом, производить пусконаладочные работы, индивидуальные испытании и комплексное опробование агрегатов, механизмов и устройств.
2. Под комплектно-блочным методом монтажа понимается организация монтажа оборудования и трубопроводов с максимальным переносом работ со строительной площадки в условия промышленного производства с агрегированием оборудования, трубопроводов и конструкций в блоки на предприятиях-поставщиках, а также на сборочно-комплектовочных предприятиях строительной индустрии и строительно-монтажных организаций с поставкой на стройки в виде комплектов блочных устройств.
1.4. В документации, выдаваемой в соответствии с п. 1.2 монтажной организации, должны быть предусмотрены:
а) применение технологических блоков и блоков коммуникаций с агрегированием, их составных частей на основе номенклатуры и технических требований, утвержденных или взаимно согласованных вышестоящими организациями заказчика и подрядчика, осуществляющего монтажные работы;
б) разделение объекта строительства на технологические узлы, состав и границы которых определяет проектная организация по согласованию с заказчиком и подрядчиком, осуществляющим монтажный работы;
в) возможность подачи технологических блоков и блоков коммуникаций к месту монтажа в собранном виде с созданием в необходимых случаях монтажных проемов в стенах и перекрытиях зданий и шарнирных устройств в опорных строительных конструкциях для монтажа методом поворота, а также с усилением при необходимости строительных конструкций для восприятия ими дополнительных временных нагрузок, возникающих в процессе монтажа; постоянные или временные дороги для перемещения тяжеловесного и крупногабаритного оборудования, а также кранов большой грузоподъемности;
г) данные по допускам для расчета точности выполнения геодезических разбивочных работ и создания внутренней геодезической разбивочной основы для монтажа оборудования.
1.5. Генподрядчик должен привлекать монтажную организацию к рассмотрению и составлению заключения по проекту организации строительства, конструктивным решениям зданий и сооружений, а также технологическим компоновкам, в которых должны быть определены возможность и основные условия производства работ комплектно-блочным и узловым методами.
1.6. Генподрядчик должен обеспечить, а монтажная организация — поручить от генподрядчика (или, по согласованию с ним, непосредственно от заказчика) необходимый комплект рабочей документации с отметкой заказчика на каждом чертеже (экземпляре) о принятии к производству.
1.7. Поставку оборудования, трубопроводов и необходимых для монтажа комплектующих изделий и материалов следует осуществлять по графику, согласованному с монтажной организацией, где должна предусматриваться первоочередная поставка машин, аппаратов, арматуры, конструкций, изделий и материалов, включенных в спецификации на блоки, подлежащие изготовлению монтажными организациями.
1.8. Окончанием работ по монтажу оборудования и трубопроводов надлежит считать завершение индивидуальных испытаний, выполненных в соответствии с разд. 5 настоящих правил, и подписание рабочей комиссией акта приемки оборудования.После окончания монтажной организацией работ по монтажу, т. е. завершения индивидуальных испытаний и приемки оборудования под комплексное опробование, заказчик производит комплексное опробование оборудования в соответствии с обязательным приложением 1.
1.9. На каждом объекте строительства в процессе монтажа оборудования и трубопроводов следует вести общий и специальные журналы производства работ согласно СНиП по организации строительного производства и оформлять производственную документацию, виды и содержание которой должны соответствовать обязательному приложению 2, а ее формы — устанавливаться ведомственными нормативными документами.
Посмотреть в PDF
Скачать
Монтаж технологических трубопроводов в Москве и МО под ключ в сжатые сроки
Монтаж технологических трубопроводов на промышленном предприятии даст возможность использования газов, пара, жидкостей. Для этих целей используются трубопроводы различной конфигурации, монтаж которых выполнят специалисты компании «Синтез ТМК».
Для этих целей используются трубопроводы различной конфигурации, монтаж которых выполнят специалисты компании «Синтез ТМК».
При проектировании трубопроводов будут учтены все особенности подключаемого оборудования, конструкции здания, расположения инженерных сетей. Мы выполним все работы по монтажу трубопроводов от закупки необходимых материалов и арматуры, подготовки отверстий и креплений под трубы до подключения к агрегатам и сетям, тестирования и ввода в эксплуатацию.
Среди предлагаемых услуг:
- Проектирование и расчет трубопроводов;
- Монтаж технологических трубопроводов, в том числе внутрицеховых и межцеховых;
- Подключение к коммуникациям и магистралям.
Для перемещения жидкостей, газов и сыпучих материалов используются технологические трубопроводы, складывающиеся из отдельных отрезков труб, соединенных при помощи арматуры или сваркой, с установленными измерительными приборами, приборами, автоматикой, изоляционными материалами. В зависимости от назначения и материала изготовления труб подбираются подходящие элементы запорной арматуры, краны, прокладки.
В зависимости от назначения и материала изготовления труб подбираются подходящие элементы запорной арматуры, краны, прокладки.
Из всех трубопроводов на промышленном предприятии более 30% составляют технологические. Они обеспечивают технологический процесс, транспортируя вещество как потребителям в качестве сырья или полуфабрикатов, так и от них, представляя собой готовую продукцию, отходы производства. Трубопроводы используются для транспортировки токсичных, вредных для здоровья, пожаро- и взрывоопасных веществ, а также жидкостей и пара, имеющих высокую температуру. Способы монтажа для различных технологических трубопроводов могут различаться, на что влияет ряд факторов.
Материал изготовления трубопроводов может быть различным, но решение, применять черные или цветные металлы, пластмассы или стекло, зависит от переносимой среды и ее температуры, давления и других показателей. В промышленных цехах монтаж трубопроводов может занимать более 50% от всех выполняемых монтажных работ. Затрудняет укладку труб необходимость работать с тоннелями, лотками, эстакадами. Работы могут проводиться в узких и неудобных местах цехов или на большой высоте под открытым небом. Номенклатура применяемых для монтажа трубопроводов деталей и узлов отличается разнообразием разметов и конструкций.
Затрудняет укладку труб необходимость работать с тоннелями, лотками, эстакадами. Работы могут проводиться в узких и неудобных местах цехов или на большой высоте под открытым небом. Номенклатура применяемых для монтажа трубопроводов деталей и узлов отличается разнообразием разметов и конструкций.
Технология монтажа трубопроводов
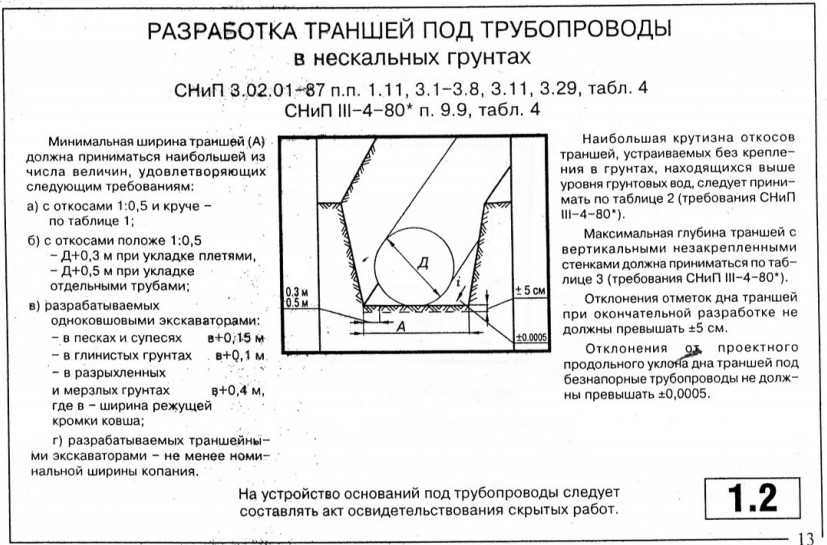
Монтаж технологических трубопроводов следует выполнять в соответствии со строительными нормами и правилами СНиП 3.05.05-84, в которых указаны основные положения производства и приемки работ по монтажу постоянных технологических трубопроводов из углеродистых и легированных сталей, цветных металлов и сплавов, чугуна, пластических масс и стекла, работающих при абсолютном давлении от 35 мм рт. ст. до 700 кгс/см2.
Объем работ по их монтажу составляет обычно около 50% общего объема монтажных работ. Прокладка большинства трубопроводов ведется в стесненных условиях, на различной высоте в многоэтажных зданиях и на открытых площадках, эстакадах, в лотках, туннелях. Внутрицеховые технологические трубопроводы отличаются большим количеством применяемых типоразмеров, деталей трубопроводов, запорно-регулирующей арматуры, средств крепления.
Внутрицеховые технологические трубопроводы отличаются большим количеством применяемых типоразмеров, деталей трубопроводов, запорно-регулирующей арматуры, средств крепления.
Классификация технологических трубопроводов
Заказчик может не вникать в сложности прокладки трубопроводов по территории цеха или через заводские площади. Для него важно выполнение функциональных задач и обеспечение станков и агрегатов газом, водой, паром. Но при этом желательно, чтобы уложенные трубы занимали минимальное место в цеху, а их прокладка не повредила существующие конструкции.
Если работы выполняет бригада с недостаточной квалификацией, в процессе монтажа трубопровода могут возникнуть непредвиденные сложности, что вызовет задержку работ, согласование с проектировщиком, дополнительные расходы.
Монтаж трубопроводов предусматривает:
- разработка ППР;
- проведение подготовительных работ;
- поставку материалов и оборудования;
- сборку, установку в проектное положение и крепление технологического трубопровода, подсоединение его к оборудованию или коммуникациям;
- установку компенсаторов
- заключительные работы по монтажу и сдаче трубопроводов;
- пуско-наладочные работы
- контроль выполнения работ
- испытания
- ведения лабораторного и геодезического контроля
- ведение исполнительной документации
Наша компания обладает достаточным опытом проведения монтажных работ, что позволяет должным образом выполнить все этапы. Если монтаж производится по готовому проекту, все пункты будут проанализированы, и при необходимости будут предложены и согласованы некоторые изменения, что позволит повысить эффективность работ.
Если монтаж производится по готовому проекту, все пункты будут проанализированы, и при необходимости будут предложены и согласованы некоторые изменения, что позволит повысить эффективность работ.
Опытные сотрудники компании выполняют контроль качества как поступающих на сборку материалов и комплектующих, так и выполнения каждой операции и конечного результата. Все стыки и швы будут выполнены аккуратно и качественно. Трубопровод будет подключен к внешним сетям, проведен тестовый пуск, получены необходимые разрешения.
При подготовительных работах к монтажу трубопроводов следует:
- обеспечить поставку и приемку в монтаж технологических материалов и оборудования;
- выполнить входной контроль проектной документации;
- выполнить входной контроль поставленных трубопроводов и оборудования, креплений и фитингов для монтажа трубопроводов;
- проверка всех типов соединений монтируемых трубопроводов к технологическому оборудованию;
- выполнить проверку технической документации;
- обеспечить хранение материалов и оборудования;
- провести сборку элементов трубопроводов;
- проверить готовность строительных конструкций и помещений сдаваемых под монтаж трубопроводов;
- проверить наличие согласований всех мест с пересечениями других инженерных систем.


Наши специалисты имеют необходимые для ведения работ сертификаты и допуски. Основным преимуществом сотрудничества с нашей компанией является возможность решения различных задач, что позволяет любые проблемы при монтажных работах решать быстро и с минимальными затратами. А если выполнение монтажа трубопроводов является частью крупного проекта, мы готовы взяться за выполнение комплекса задач, обеспечив оптимальные условия для получения превосходного результата.
Монтаж технологических трубопроводов регулируется следующими нормативными документами:
СНиП 3.05.05-84 Технологическое оборудование и технологические трубопроводы
ВСН 362-87 Изготовление, монтаж и испытания технологических трубопроводов до 10 МПА
ВСН 70-79 Инструкция по монтажу и испытанию трубопроводов диаметром условного прохода до 400 мм включительно на давление свыше 9.8 до 245 МПА
СН 527-80 Инструкция по проектированию стальных трубопроводов до 10 МПА
ГОСТ 21.401-88 Система проектной документации для строительства. Технология производства. Основные требования к рабочим чертежам
Технология производства. Основные требования к рабочим чертежам
Сборник Е26 Монтаж технологических трубопроводов
Поделиться:
Бесплатная консультация
Нажимая кнопку “отправить”, вы соглашаетесь с политикой конфиденциальности
узнать стоимость работ
ngs_backbone: конвейер для очистки чтения, сопоставления и вызова SNP с использованием последовательности следующего поколения | BMC Genomics
- Программное обеспечение
- Открытый доступ
- Опубликовано:
- Хосе М. Бланка 1 ,
- Лаура Паскуаль 1 ,
- Пейо Зиарсоло 1 ,
- Фернандо Нуэс 1 и
- …
- Хоакин Каньисарес 1
БМС Геномика том 12 , Номер статьи: 285 (2011) Процитировать эту статью
14 000 обращений
48 цитирований
1 Альтметрический
Сведения о показателях
Abstract
Background
Возможности платформ секвенирования нового поколения (NGS) произвели революцию в биотехнологических лабораториях. Более того, сочетание NGS-секвенирования и доступных высокопроизводительных технологий генотипирования способствует быстрому обнаружению и использованию SNP у немодельных видов. Однако это изобилие последовательностей и полиморфизмов создает новые потребности в программном обеспечении. Чтобы удовлетворить эти потребности, мы разработали мощное, но простое в использовании приложение.
Более того, сочетание NGS-секвенирования и доступных высокопроизводительных технологий генотипирования способствует быстрому обнаружению и использованию SNP у немодельных видов. Однако это изобилие последовательностей и полиморфизмов создает новые потребности в программном обеспечении. Чтобы удовлетворить эти потребности, мы разработали мощное, но простое в использовании приложение.
Результаты
Программное обеспечение ngs_backbone представляет собой параллельный конвейер, способный анализировать считывания последовательностей Sanger, 454, Illumina и SOLiD (секвенирование путем лигирования и обнаружения олигонуклеотидов). Его основными поддерживаемыми анализами являются: очистка чтения, сборка и аннотация транскриптома, картирование чтения и вызов и выбор полиморфизма одиночных нуклеотидов (SNP). Чтобы создать действительно полезный инструмент, разработка программного обеспечения была сопряжена с лабораторным экспериментом. Для тестирования инструмента и прогнозирования полиморфизма томатов были использованы все общедоступные чтения томатов Sanger EST плюс 14,2 миллиона прочтений Illumina. Очищенные чтения были сопоставлены с транскриптомом томата SGN, получив покрытие 4,2 для Sanger и 8,5 для Illumina. Было предсказано 23 360 однонуклеотидных вариаций (SNV). Всего было экспериментально подтверждено 76 SNV, из которых 85% оказались реальными.
Очищенные чтения были сопоставлены с транскриптомом томата SGN, получив покрытие 4,2 для Sanger и 8,5 для Illumina. Было предсказано 23 360 однонуклеотидных вариаций (SNV). Всего было экспериментально подтверждено 76 SNV, из которых 85% оказались реальными.
Выводы
ngs_backbone — это новый программный пакет, способный анализировать последовательности, созданные технологиями NGS, и предсказывать SNV с высокой точностью. В нашем примере с помидорами мы создали высокополиморфную коллекцию SNV, которая будет полезным ресурсом для исследователей и селекционеров томатов. Программное обеспечение, разработанное вместе с документацией, находится в свободном доступе по лицензии AGPL и может быть загружено с http://bioinf.comav.upv.es/ngs_backbone/ или http://github.com/JoseBlanca/franklin.
Исходная информация
Возможности, предлагаемые платформами секвенирования следующего поколения (NGS), революционизируют биотехнологические лаборатории, но для реализации их истинного потенциала еще предстоит преодолеть одно препятствие: анализ данных [1]. В настоящее время получение последовательности de novo , 454 (454 Life Sciences, Roche. Branford, CT, USA[2]) для транскриптома немодельного вида или последовательности на основе Illumina (Illumina, Сан-Диего, Калифорния, США [3]) геномное или транскриптомное повторное секвенирование нескольких образцов очень доступно.
В настоящее время получение последовательности de novo , 454 (454 Life Sciences, Roche. Branford, CT, USA[2]) для транскриптома немодельного вида или последовательности на основе Illumina (Illumina, Сан-Диего, Калифорния, США [3]) геномное или транскриптомное повторное секвенирование нескольких образцов очень доступно.
Однако эти новые технологии секвенирования нельзя анализировать с помощью более старого программного обеспечения, разработанного для секвенирования по Сэнгеру. И количество, и качество данных сильно различаются [1, 4]. Постоянно создается множество новых программ и форматов данных: ассемблеры (Mira [5], Newbler (454 Life Sciences, Roche. Branford, CT, USA[2]), картографы (например, bwa [6], Bowtie [7 ]) и форматы файлов (SAMtools [8], VCF [9]). Эти стремительные разработки сделали область биоинформатики очень динамичной и, следовательно, трудной для понимания, несмотря на рекомендации, предоставленные такими ресурсами, как интернет-форум SEQanswers [10], который посвящен представлению и документированию инструментов, используемых для анализа данных NGS. Кроме того, после выбора оптимальных инструментов и завершения анализа получаются огромные файлы. Эти файлы обычно обрабатываются путем создания небольших частей программного обеспечения, называемых сценариями. По нашему мнению, как выбор различных программ и параметров, так и создание этих небольших скриптов делают процесс анализа громоздким и невоспроизводимым, особенно если в лаборатории нет специального персонала по биоинформатике.
Кроме того, после выбора оптимальных инструментов и завершения анализа получаются огромные файлы. Эти файлы обычно обрабатываются путем создания небольших частей программного обеспечения, называемых сценариями. По нашему мнению, как выбор различных программ и параметров, так и создание этих небольших скриптов делают процесс анализа громоздким и невоспроизводимым, особенно если в лаборатории нет специального персонала по биоинформатике.
Эти проблемы можно решить, используя стандартизированный метод и простые в использовании конвейеры, способные комплексно анализировать все шаги, необходимые для перехода от необработанных последовательностей NGS к набору окончательных аннотаций. Некоторые заметные предыдущие усилия в этой области привели к созданию нескольких трубопроводов. Тремя яркими примерами являются Galaxy [11], CloVR [12] и est2assembly [13]. est2assembly — хороший сборочный конвейер, но, к сожалению, он не умеет обрабатывать чтения Illumina и SOLiD (Sequencing by Oligoнуклеотид Ligation and Detection) [14], не работает параллельно и не делает сопоставление чтений. Применение платформ секвенирования NGS безгранично [15–18]. Например, до недавнего времени использование SNP у немодельных видов было редкостью. Однако сочетание NGS-секвенирования и доступных высокопроизводительных технологий генотипирования (таких как Massarray (Sequenom, Сан-Диего, Калифорния, США), Veracode или BeadArray (Illumina, Сан-Диего, Калифорния, США)[3] способствует быстрому обнаружению и использование этих молекулярных маркеров. Миллионы последовательностей теперь могут быть сгенерированы с низкими затратами, и, учитывая простой способ их анализа, можно быстро получить огромное количество SNP. Такое изобилие SNP создает потребность в новом программном обеспечении, поскольку исследователи обычно заинтересованы в выборе подмножества SNP, предназначенного для конкретного эксперимента, SNP в этих подмножествах должны иметь низкий уровень ошибок, должны быть адаптированы к используемой методике генотипирования и должны быть вариабельными у индивидуумов, подлежащих генотипированию.
Применение платформ секвенирования NGS безгранично [15–18]. Например, до недавнего времени использование SNP у немодельных видов было редкостью. Однако сочетание NGS-секвенирования и доступных высокопроизводительных технологий генотипирования (таких как Massarray (Sequenom, Сан-Диего, Калифорния, США), Veracode или BeadArray (Illumina, Сан-Диего, Калифорния, США)[3] способствует быстрому обнаружению и использование этих молекулярных маркеров. Миллионы последовательностей теперь могут быть сгенерированы с низкими затратами, и, учитывая простой способ их анализа, можно быстро получить огромное количество SNP. Такое изобилие SNP создает потребность в новом программном обеспечении, поскольку исследователи обычно заинтересованы в выборе подмножества SNP, предназначенного для конкретного эксперимента, SNP в этих подмножествах должны иметь низкий уровень ошибок, должны быть адаптированы к используемой методике генотипирования и должны быть вариабельными у индивидуумов, подлежащих генотипированию.
Мы стремились создать мощное, но простое в использовании приложение, способное выполнять анализ последовательности NGS и прогнозирование полиморфизма. Чтобы создать действительно полезный инструмент, разработка программного обеспечения была сопряжена с лабораторным экспериментом: поиском SNV в томате. У этого вида из-за узкой генетической базы [19–21] оказалось трудно найти высокополиморфные SNP. В нескольких предыдущих исследованиях были изучены общедоступные базы данных EST томатов [22–25]. В этих исследованиях были предсказаны тысячи SNP томата между определенными образцами. К сожалению, полиморфизм этих SNP в других образцах томатов не оценивался и не сообщался, что делает эти SNP громоздкими для использования в других материалах томата. Другие подходы позволили найти SNP томата и сообщить об их полиморфизме, но только несколько сотен SNP были получены с помощью ручного повторного секвенирования [26] и гибридизации массива олигонуклеотидов [27]. Поиск новых и высокоинформативных SNV (однонуклеотидных вариаций, SNP плюс вставки) путем повторного секвенирования транскриптома томата сам по себе будет чрезвычайно полезен для ученых и селекционеров, работающих над этим видом.
Программное обеспечение, разработанное вместе с его документацией, находится в свободном доступе по лицензии AGPL и может быть загружено с веб-сайта службы биоинформатики COMAV [28], а также в дополнительном файле 1.
Реализация
Когда архитектура ngs_backbone была Созданный, несколько характеристик считались важными: использование стандартных форматов файлов и сторонних бесплатных программных инструментов, модульность и расширяемость, воспроизводимость анализа и простота использования.
Для облегчения взаимодействия с другими инструментами большинство входных и выходных файлов имеют стандартный формат, например FASTA, FASTQ, BAM, VCF и GFF, который может создаваться и использоваться другими инструментами. Например, очень легко просмотреть сопоставление и аннотацию, полученные путем загрузки файлов BAM и GFF в программу просмотра, такую как IGV [29].
ngs_backbone использует сторонние инструменты признанного качества, такие как SAMtools или GATK, когда это возможно, для поддержания качества анализа. Такой подход сказывается на процессе установки, но чтобы сделать его менее сложным, мы упаковали и предварительно скомпилировали большинство этих сторонних инструментов и написали подробное пошаговое руководство по установке, которое распространяется вместе с инструментом [30]. ].
Такой подход сказывается на процессе установки, но чтобы сделать его менее сложным, мы упаковали и предварительно скомпилировали большинство этих сторонних инструментов и написали подробное пошаговое руководство по установке, которое распространяется вместе с инструментом [30]. ].
Модульность также была важной целью архитектуры ngs_backbone. Пользователи требуют проведения постоянно меняющегося набора анализов, и эти анализы необходимо корректировать для каждого проекта. Чтобы удовлетворить это требование, был создан набор функций сопоставления, ориентированных на различные задачи, такие как очистка или аннотирование. Эти функции имеют общий интерфейс, все они берут последовательность и генерируют новую, измененную последовательность и составляют шаги наших пайплайнов, которые генерируются во время выполнения для каждого анализа.
Наконец, несмотря на то, что мы представляем ngs_backbone как инструмент командной строки, это не единственный способ его использования. Базовая библиотека, на которой работает этот инструмент, называется franklin и написана на Python. У этой библиотеки есть другие возможности, которые в настоящее время не доступны через существующий интерфейс командной строки, но ее API задокументирован и прост в использовании, и программисты Python, желающие разрабатывать свои собственные сценарии и инструменты поверх нее, могут это сделать. За его развитием можно следить на сайте github [31], и его лицензия также открыта (AGPL).
У этой библиотеки есть другие возможности, которые в настоящее время не доступны через существующий интерфейс командной строки, но ее API задокументирован и прост в использовании, и программисты Python, желающие разрабатывать свои собственные сценарии и инструменты поверх нее, могут это сделать. За его развитием можно следить на сайте github [31], и его лицензия также открыта (AGPL).
Результаты и обсуждение
Алгоритмы конвейера ngs_backbone
В этом разделе описываются методы, используемые внутри ngs_backbone. Упомянутое стороннее программное обеспечение не должно запускаться пользователем, поскольку оно будет использоваться только внутри ngs_backbone. Всего пары команд (backbone_create_project и backbone_analyze) будет достаточно для завершения любого анализа.
Типичный анализ, выполняемый ngs_backbone, начинается с набора файлов чтения Sanger, 454, Illumina или SOLiD. Первым шагом является очистка чтения. В этом процессе адаптер, вектор и некачественные области удаляются. Точный алгоритм, используемый для каждого шага очистки, зависит от типа чтения. Например, обрезка качества в длинных чтениях выполняется Люси [32], но для более коротких чтений вместо этого используется внутренний алгоритм. Дополнительные сведения о множестве доступных модулей очистки чтения см. в документации, распространяемой вместе с инструментом [28]. После завершения очистки можно создать распределения качества и длины для необработанных и чистых показаний в качестве оценки качества процесса очистки.
Точный алгоритм, используемый для каждого шага очистки, зависит от типа чтения. Например, обрезка качества в длинных чтениях выполняется Люси [32], но для более коротких чтений вместо этого используется внутренний алгоритм. Дополнительные сведения о множестве доступных модулей очистки чтения см. в документации, распространяемой вместе с инструментом [28]. После завершения очистки можно создать распределения качества и длины для необработанных и чистых показаний в качестве оценки качества процесса очистки.
Если эталонный транскриптом недоступен, его можно собрать с помощью чистых прочтений с помощью ассемблера MIRA [5]. MIRA позволяет использовать гибридные сборки со считываниями Sanger, 454 и Illumina. ngs_backbone автоматизирует подготовку проекта MIRA. После запуска MIRA полученный набор контигов может быть аннотирован всеми доступными аннотациями: микросателлит, ORF, функциональное описание, термины GO, расположение интронов и ортологи.
После того, как референсный транскриптом или геном будет доступен, чтения могут быть сопоставлены с ним. Используемый для отображения алгоритм также зависит от длины считывания. Короткие чтения отображаются стандартным алгоритмом bwa [6], в то время как более длинные чтения используют алгоритм BWT-SW. ngs_backbone создает BAM-файл со всеми сопоставленными операциями чтения. Сгенерированные файлы BAM обрабатываются с помощью SAMtools [8] и Picard [33], объединяются и адаптируются для GATK [34] с запуском пользовательского кода.
Используемый для отображения алгоритм также зависит от длины считывания. Короткие чтения отображаются стандартным алгоритмом bwa [6], в то время как более длинные чтения используют алгоритм BWT-SW. ngs_backbone создает BAM-файл со всеми сопоставленными операциями чтения. Сгенерированные файлы BAM обрабатываются с помощью SAMtools [8] и Picard [33], объединяются и адаптируются для GATK [34] с запуском пользовательского кода.
Одной из частых целей проектов, использующих последовательности NGS, является поиск вариаций последовательности (SNP и небольшие вставки). Чтобы улучшить этот процесс, перед вызовом SNV можно выполнить повторное выравнивание файла BAM с помощью GATK [34]. Для вызова SNV операции чтения с качеством отображения ниже 15 не учитываются. Аллельные качества рассчитываются с использованием качества трех наиболее надежных прочтений (PQ1, PQ2, PQ2) по формуле PQ1 + 0,25 * (PQ2 + PQ3). Этот метод представляет собой небольшую вариацию метода, используемого MIRA [5] для расчета качества согласованной позиции. Аннотация SNV учитывает накопленное качество последовательности для каждого аллеля, а также качество картирования для каждого чтения. Для обоих параметров устанавливается пороговое значение, и только позиции с двумя или более качественными аллелями считаются SNP или вставками. Из этих файлов BAM обычно генерируются тысячи SNV, поэтому для возможности выбора наиболее полезных был разработан набор фильтров SNV (таблица 1). Код, используемый для запуска фильтров SNV, полностью был написан для ngs_backbone. Окончательно полученные SNV вместе с информацией о фильтре сохраняются в файле VCF [9].].
Аннотация SNV учитывает накопленное качество последовательности для каждого аллеля, а также качество картирования для каждого чтения. Для обоих параметров устанавливается пороговое значение, и только позиции с двумя или более качественными аллелями считаются SNP или вставками. Из этих файлов BAM обычно генерируются тысячи SNV, поэтому для возможности выбора наиболее полезных был разработан набор фильтров SNV (таблица 1). Код, используемый для запуска фильтров SNV, полностью был написан для ngs_backbone. Окончательно полученные SNV вместе с информацией о фильтре сохраняются в файле VCF [9].].
Полноразмерная таблица
Несмотря на то, что описанный анализ является типичным, каждый из шагов на самом деле является необязательным. Конвейер имеет несколько точек входа и результатов. Можно начать с необработанных последовательностей и выполнить только очистку или же начать с файла BAM и использовать инструмент для вызова SNV. Каждый анализ независим от других; он просто принимает набор стандартных файлов в качестве входных данных и генерирует другой набор стандартных файлов в качестве выходных данных.
Каждый анализ независим от других; он просто принимает набор стандартных файлов в качестве входных данных и генерирует другой набор стандартных файлов в качестве выходных данных.
Анализ томатов ngs_backbone с помощью программного обеспечения
Для тестирования инструмента был проведен полный анализ транскриптома помидоров, от очистки считывания до экспериментальной проверки SNV. Все эти анализы проводились с использованием ngs_backbone. В это исследование были включены все общедоступные чтения Sanger EST томатов, доступные в базах данных SGN и GenBank [35, 36] с известным происхождением образцов томатов. В дополнение к этим последовательностям Сэнгера, 14,2 миллиона прочтений Illumina, полученных из нормализованной библиотеки кДНК, построенной с использованием эквимолярной смеси цветочной РНК, выделенной из линий томатов UC-82 и RP75/79., были добавлены (дополнительный файл 2).
После удаления некачественных участков и загрязнений вектора и адаптера осталось 9,8 миллиона последовательностей Illumina и 276 039 последовательностей Сэнгера. Наиболее представленными линиями томатов были Micro Tom (118 304 последовательности), TA496 (104 503 последовательности) и смесь RP75/59-UC82 (9,8 миллиона последовательностей). ngs_backbone рассчитал статистику о функциях последовательности и процессе очистки (рис. 1).
Наиболее представленными линиями томатов были Micro Tom (118 304 последовательности), TA496 (104 503 последовательности) и смесь RP75/59-UC82 (9,8 миллиона последовательностей). ngs_backbone рассчитал статистику о функциях последовательности и процессе очистки (рис. 1).
Статистический анализ ngs_backbone . Распределение длин очищенных последовательностей Сэнгера (а) и Иллюмины (б). Блочная диаграмма лекции о качестве парного основания относительно положения последовательности последовательностей Сэнгера (c) и Illumina (d). Распределение покрытия последовательностей выравнивания последовательностей Sanger (e) и Illumina (f).
Изображение полного размера
Очищенные чтения были картированы на транскриптоме томата SGN [35]. Было нанесено на карту 7,75 миллиона прочтений Illumina, а также все чтения Сэнгера, что позволило получить среднее покрытие 4,2 для последовательностей Сэнгера и 8,5 для последовательностей Illumina (рис. 1). Чтобы улучшить это выравнивание, функциональность повторного выравнивания, предоставляемая GATK [34], была применена до вызова SNV.
1). Чтобы улучшить это выравнивание, функциональность повторного выравнивания, предоставляемая GATK [34], была применена до вызова SNV.
В аннотации SNV учитывалось накопленное качество последовательности для каждого аллеля, а также качество картирования для каждого считывания. Для обоих параметров был установлен порог, и только позиции с двумя или более качественными аллелями считались SNP или вставками. Все 33 306 найденных SNV заносятся в файл VCF (дополнительный файл 3).
Несмотря на соответствие критериям качества, не все SNV оказались одинаково надежными. Было применено несколько фильтров, чтобы пометить тех, кто с наибольшей вероятностью был реальным (таблица 1). Например, к набору Illumina был применен фильтр частоты наиболее частых аллелей (MAF), поскольку в большинстве случаев при смешивании двух эквимолярных образцов кДНК ожидается соотношение между аллелями, близкое к 0,5. В нашем случае смесь соответствовала линиям томатов UC-82 и RP75/79., и ожидалось, что аллели, присутствующие в обоих из них, появятся в EST одинаковое количество раз для большинства unigenes. Кроме того, был применен фильтр, который помечал SNV в высоковариабельных областях (HVR), чтобы избежать unigenes со слишком большой изменчивостью. Считалось, что 23 360 SNV, которые прошли оба фильтра, имеют более высокую вероятность быть реальными и составили набор HL (таблица 2). Подсчеты SNV, представленные с этого момента, не будут включать SNV, которые не прошли эти фильтры, если это не указано явно.
Кроме того, был применен фильтр, который помечал SNV в высоковариабельных областях (HVR), чтобы избежать unigenes со слишком большой изменчивостью. Считалось, что 23 360 SNV, которые прошли оба фильтра, имеют более высокую вероятность быть реальными и составили набор HL (таблица 2). Подсчеты SNV, представленные с этого момента, не будут включать SNV, которые не прошли эти фильтры, если это не указано явно.
Полноразмерная таблица
При использовании SNV в экспериментальных условиях не все одинаково полезны и просты в использовании. В зависимости от метода, используемого для их обнаружения, некоторые характеристики SNV могут облегчить или затруднить эксперимент, например близость к границе интрона, к другому SNV или к концу унигена. Кроме того, SNV, расположенные в unigenes, которые очень похожи на другие unigenes, были помечены, чтобы избежать семейств генов, которые могли затруднить отслеживание процессов ПЦР и конструирования праймеров. Это было сделано путем применения фильтров Unique и Continuous Region, I30 и CL30, доступных в коллекции фильтров ngs_backbone (таблица 1). Все фильтры, примененные для маркировки SNV, а также полученные результаты показаны в таблице 3. 6,934 SNV, прошедших эти фильтры, составили легко используемый набор (EU).
Это было сделано путем применения фильтров Unique и Continuous Region, I30 и CL30, доступных в коллекции фильтров ngs_backbone (таблица 1). Все фильтры, примененные для маркировки SNV, а также полученные результаты показаны в таблице 3. 6,934 SNV, прошедших эти фильтры, составили легко используемый набор (EU).
Полноразмерная таблица
Также желательно пометить SNV с высоким полиморфизмом.
Основное преимущество этих высокополиморфных маркеров заключается в простоте их использования у разных людей. SNV с низким PIC (полиморфным информационным содержанием) имеют низкую вероятность наличия разных аллелей между двумя случайно выбранными людьми. Обогащая выборку высокополиморфными SNV, увеличивается доля различающих SNV в любом эксперименте, имеющем дело со случайным набором особей, что снижает затраты на лабораторию.
Полиморфизм в популяции может быть правильно выведен только при наличии обширной и хорошо генотипированной выборки особей. Поскольку EST передают информацию о генотипе от разных людей, ngs_backbone делает грубую оценку полиморфизма для каждого SNV, подсчитывая количество образцов томатов, в которых появляется каждый аллель. Надежность этого предполагаемого полиморфизма зависит, среди прочего, от количества секвенированных индивидуумов. Принимая во внимание только SNV, секвенированные как минимум в шести различных образцах томата, 514 SNV с частотой наиболее распространенного аллеля менее 0,67 были включены в полиморфный (PO) набор. Этот набор был небольшим, несмотря на хороший охват последовательностей для четырех образцов томатов, поскольку из других материалов томатов было доступно не так много последовательностей. Пересечение этого набора ПО с легко используемым (ЕС) дало 291 СНВ.
Поскольку EST передают информацию о генотипе от разных людей, ngs_backbone делает грубую оценку полиморфизма для каждого SNV, подсчитывая количество образцов томатов, в которых появляется каждый аллель. Надежность этого предполагаемого полиморфизма зависит, среди прочего, от количества секвенированных индивидуумов. Принимая во внимание только SNV, секвенированные как минимум в шести различных образцах томата, 514 SNV с частотой наиболее распространенного аллеля менее 0,67 были включены в полиморфный (PO) набор. Этот набор был небольшим, несмотря на хороший охват последовательностей для четырех образцов томатов, поскольку из других материалов томатов было доступно не так много последовательностей. Пересечение этого набора ПО с легко используемым (ЕС) дало 291 СНВ.
Чтобы увеличить количество предполагаемых высокополиморфных SNV, были также применены менее строгие критерии, создав новый набор с вариабельными SNV как в последовательностях Illumina, так и в последовательностях Sanger, независимо от их предполагаемого полиморфизма. Было отобрано 2855 SNV, из которых 860 также присутствовали в отборе ЕС (таблица 3). Эти SNV были названы обычными (CO), поскольку они были полиморфными в общедоступной коллекции EST, а также в последовательностях Illumina. SNV, которые оказались полиморфными только в коллекциях Sanger или Illumina, были названы SA и IL соответственно.
Было отобрано 2855 SNV, из которых 860 также присутствовали в отборе ЕС (таблица 3). Эти SNV были названы обычными (CO), поскольку они были полиморфными в общедоступной коллекции EST, а также в последовательностях Illumina. SNV, которые оказались полиморфными только в коллекциях Sanger или Illumina, были названы SA и IL соответственно.
Экспериментальная проверка прогнозов программного обеспечения
Качество вызова in silico SNV было протестировано в коллекции из 37 образцов томатов, включающих 10 коммерческих сортов и 27 местных сортов томатов (дополнительный файл 4). Для генотипирования этих материалов использовался метод HRM PCR (ПЦР с плавлением высокого разрешения) [37]. Чтобы сопоставить кривые плавления с аллелями SNV, образцы RP75/59 и UC-82, входящие в набор Illumina EST, по возможности использовали в качестве контроля. Когда между этими образцами не ожидалось полиморфизма, для дифференциации аллелей использовали полиморфизм рестрикционных ферментов (также предсказанный ngs_backbone).
Всего было экспериментально протестировано 76 in silico SNV (дополнительные файлы 2, 5). Техника HRM смогла подтвердить 85% из них (таблица 4). Этот высокий уровень успеха делает возможным использование предсказанных in silico SNV даже без какой-либо предварительной обширной экспериментальной проверки. Более того, вероятность успеха была, по всей вероятности, занижена из-за использованной методики эксперимента. HRM-ПЦР не способна различить все пары аллелей, и вполне вероятно, что в некоторых случаях невозможность обнаружения некоторых из in silico — предсказанные SNV были вызваны ошибкой в PCR.
Таблица 4. Статистические данные для проанализированных SNV в различных коллекциях.Полноразмерная таблица
Такой высокий уровень успеха был достигнут, несмотря на низкий охват (4,2 для Sanger и 8,5 для Illumina), хотя, вероятно, он был получен за счет низкой специфичности, которая не оценивалась в представленном экспериментальном плане.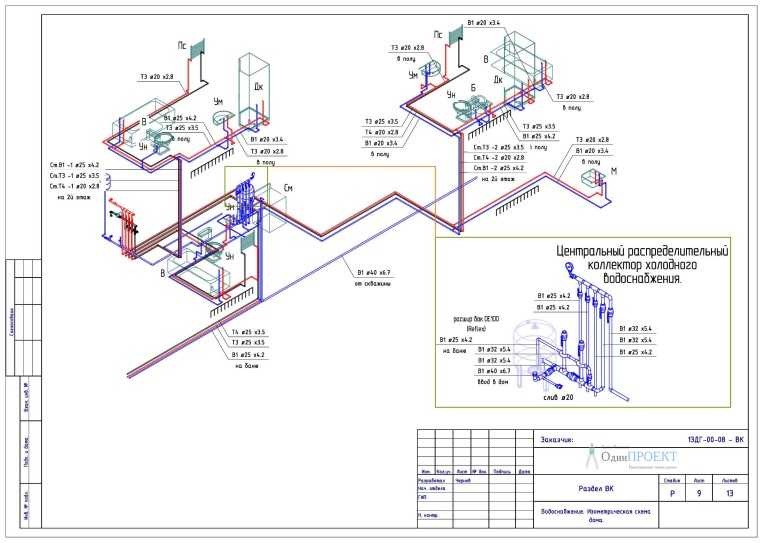 Параметры, используемые для отбора, были даже скорректированы таким образом, чтобы пометить как ненадежные некоторые SNV, которые, даже если они были подтверждены достаточным охватом, находились в регионах с высокой изменчивостью или имели несбалансированную частоту аллеля в эквимолярном RP75/. 59- Образец UC-82 Illumina.
Параметры, используемые для отбора, были даже скорректированы таким образом, чтобы пометить как ненадежные некоторые SNV, которые, даже если они были подтверждены достаточным охватом, находились в регионах с высокой изменчивостью или имели несбалансированную частоту аллеля в эквимолярном RP75/. 59- Образец UC-82 Illumina.
Одной из целей этого исследования было разработать и протестировать стратегию выбора наиболее полиморфного подмножества SNV с использованием как общедоступных, так и новых тестов Illumina EST. Хотя невозможно сделать точную оценку PIC, просто используя набор общедоступных последовательностей, собранных из разных гетерогенных проектов, приблизительный индекс, связанный с полиморфизмом, можно рассчитать, подсчитав количество людей, у которых появляется каждый аллель. Несмотря на несколько искажающих факторов, низкий PIC SNV будет иметь тенденцию к очень несбалансированному индивидуальному подсчету различных аллелей. Ожидаемый средний полиморфизм наборов SVN с разными PIC оценивали путем генотипирования 37 образцов томатов. Два набора SNV использовались для определения ожидаемого базового уровня полиморфизма. Единственным фильтром, связанным с полиморфизмом, применяемым к этим наборам, было требование наличия по крайней мере двух разных аллелей в последовательностях Illumina или Sanger. После генотипирования коллекции томатов с использованием SNV, случайно выбранных из этих наборов, мы обнаружили, что 3 из 14 SNV, протестированных в наборе Sanger, и 5 из 12 в наборе Illumina были полиморфными, то есть наиболее часто встречающаяся частота аллеля было ниже 95%.
Два набора SNV использовались для определения ожидаемого базового уровня полиморфизма. Единственным фильтром, связанным с полиморфизмом, применяемым к этим наборам, было требование наличия по крайней мере двух разных аллелей в последовательностях Illumina или Sanger. После генотипирования коллекции томатов с использованием SNV, случайно выбранных из этих наборов, мы обнаружили, что 3 из 14 SNV, протестированных в наборе Sanger, и 5 из 12 в наборе Illumina были полиморфными, то есть наиболее часто встречающаяся частота аллеля было ниже 95%.
Другие наборы SNV, которые, как ожидалось, будут несколько более полиморфными, были созданы путем просеивания SNV, которые были полиморфны как в последовательностях Sanger, так и в последовательностях Illumina (набор CO), а также наборы из набора PO (где последовательности из по крайней мере 6 растения были доступны, и количество аллелей было достаточно сбалансированным). В обоих наборах 70% протестированных маркеров были полиморфными, что было явно выше, чем 21% и 42%, обнаруженные в исходном полиморфизме.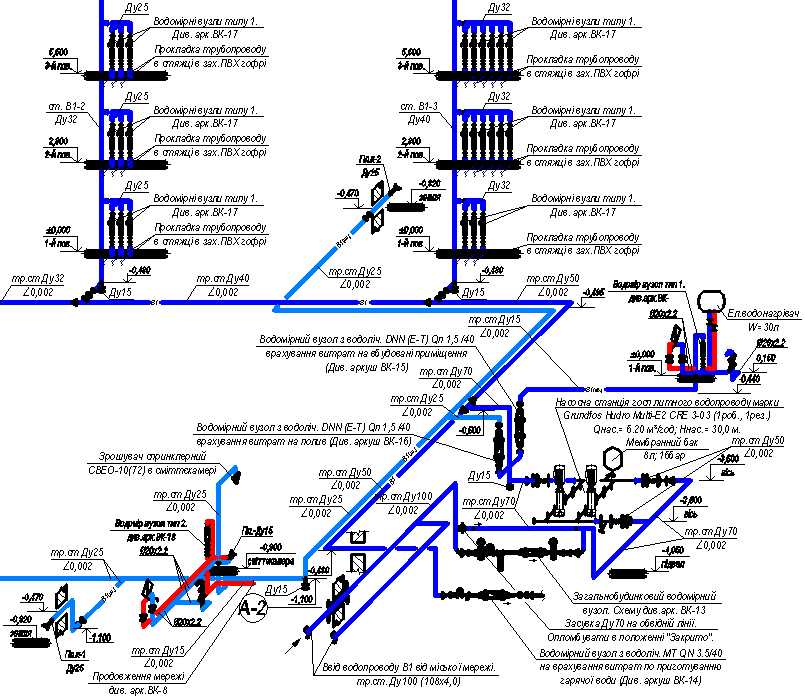
В этих наборах содержание полиморфной информации (PIC) также должно было быть выше, чем в наборах Sanger и Illumina, где PIC составлял 0,04 и 0,08 соответственно. В наборе CO PIC был фактически выше, 0,22. Наконец, SNV, которые, как ожидается, будут наиболее полиморфными, были из набора PO. В них были доступны последовательности как минимум 6 растений, и количество аллелей было вполне сбалансированным. Найденный PIC в этом случае составил 0,28, поэтому при поиске высокополиморфных SNV эта окончательная стратегия окупается.
К сожалению, подобный отбор не может быть сделан напрямую для всех немодельных видов с общедоступными EST, так как во многих случаях почти все последовательности исходят от нескольких разных особей. На самом деле, даже в публичных томатных последовательностях нет большого разнообразия. 81% этих общедоступных последовательностей были получены всего от 2 человек. Учитывая показанные результаты, мы рекомендуем при поиске SNV учитывать количество секвенированных лиц.
Заключение
Для анализа данных транскриптома NGS мы разработали модульный и параллельный конвейер с широкими возможностями настройки, написанный на Python, под названием ngs_backbone. Это программное обеспечение представляет собой новую стратегию использования технологий NGS, которая ускорит исследования немодельных видов и облегчит использование этих технологий лабораториями со специализированным персоналом в области биоинформатики или без него.
В представленном примере с помидорами анализ начался с 14,5 миллионов прочтений, которые после очистки и картирования дали 23 360 предполагаемых SNP и вставок (SNV).
Согласно экспериментальной проверке, 85% SNV, предсказанных in silico , были реальными. Этот высокий уровень успеха делает возможным использование предсказанных in silico SNV даже без какой-либо предварительной обширной экспериментальной проверки. Кроме того, коллекция из 2855 созданных высокополиморфных SNV будет полезным ресурсом, поскольку старые и винтажные томаты имеют узкую генетическую базу, что затрудняет обнаружение полиморфных маркеров в этих материалах.
Программное обеспечение ngs_backbone обеспечивает идеальный способ проведения полного анализа последовательностей NGS, включая очистку считывания, картирование, сборку транскриптома, аннотирование и вызов SNV. Это инструмент с открытым исходным кодом, выпущенный под AGPL, написанный на Python и доступный на веб-сайте службы биоинформатики COMAV [28]. Кроме того, базовая библиотека, которая поддерживает ngs-backbone, называется franklin. За его развитием можно следить на сайте github [31]. Его API задокументирован и прост в использовании, и программисты Python, желающие разрабатывать свои собственные скрипты и инструменты поверх него, могут это сделать.
Наличие и требования
Название проекта: ngs_backbone
Домашняя страница проекта : http://bioinf.comav.upv.es/ngs_backbone/ и http://github.com/JoseBlanca/franklin
9 Операционная система: Linux
Язык программирования: Python (2.6)
Другие требования: Полный список можно найти на веб-сайте.
Лицензия: Открытый исходный код, AGPL
Ограничения на использование неакадемическими пользователями: нет
Каталожные номера
Metzker ML: Технологии секвенирования – новое поколение. Природа Обзоры Генетика. 2010, 11 (1): 31-46. 10.1038/nrg2626.
КАС Статья Google ученый
454 секвенирование. [http://www.454.com/]
Illumina Inc. [http://www.illumina.com/]
Flicek P, Birney E: Смысл считываний последовательностей: методы выравнивания и сборка (том 6, стр. S6, 2009 г.). Природные методы. 2010, 7 (6): 479-479.
КАС Статья Google ученый
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WEG, Wetter T, Suhai S: Использование ассемблера miraEST для надежной и автоматизированной сборки транскриптов мРНК и обнаружения SNP в секвенированных EST.
Геномные исследования. 2004, 14 (6): 1147-1159. 10.1101/гр.1917404.КАС Статья Google ученый
Li H, Durbin R: Быстрое и точное выравнивание коротких считываний с помощью преобразования Берроуза-Уилера. Биоинформатика. 2009, 25 (14): 1754-1760. 10.1093/биоинформатика/btp324.
КАС Статья Google ученый
Лангмид Б., Трапнелл С., Поп М., Зальцберг С.Л.: Сверхбыстрое и эффективное с точки зрения памяти сопоставление коротких последовательностей ДНК с геномом человека. Геномная биология. 2009, 10 (3):
Статья Google ученый
Ли Х., Хэндсакер Б., Высокер А., Феннелл Т., Руан Дж., Гомер Н., Март Г., Абекасис Г., Дурбин Р., Данные проекта генома П: формат выравнивания последовательности/карты и SAMtools. Биоинформатика. 2009, 25 (16): 2078-2079.
10.1093/биоинформатика/btp352.Артикул Google ученый
1000 геномов. Глубокий каталог генетической изменчивости человека. [http://1000genomes.org/wiki/doku.php?id=1000_genomes:analysis:vcf4.0]
Интернет-форум seqanswers. [http://seqanswers.com/]
Бланкенберг Д., Тейлор Дж., Шенк И., Хе Дж. Б., Чжан И., Гент М., Вирарагхаван Н., Альберт И., Миллер В., Макова К. Д., Росс Ч., Некрутенко А.: Основа для совместного анализа данных ENCODE: создание крупномасштабных анализов, удобных для биологов. Геномные исследования. 2007, 17 (6): 960-964. 10.1101/гр.5578007.
КАС Статья Google ученый
Автоматический анализ последовательности CloVR с вашего рабочего стола. [http://clovr.org/]
Папаниколау А., Стирли Р., Френч-Констант Р.
Х., Хеккель Д.Г.: Транскриптомы следующего поколения для геномов следующего поколения с использованием сборки est2. БМК Биоинформатика. 2009, 10:Google ученый
Прикладные биосистемы по жизненным технологиям. [http://www.appliedbiosystems.com/absite/us/en/home/applications-technologies/solid-next-generation-sequencing.html]
Уолл П.К., Либенс-Мак Дж., Чандербали А.С., Баракат А., Уолкотт Э., Лян Х.И., Ландхерр Л., Томшо Л.П., Ху И., Карлсон Дж.Е., Ма Х., Шустер С.К., Солтис Д.Э., Солтис П.С., Альтман Н. , dePamphilis CW: Сравнение технологий секвенирования следующего поколения для характеристики транскриптома. Геномика BMC. 2009, 10:
Google ученый
Мерчисон Э.П., Товар С., Хсу А., Бендер Х.С., Херадпур П., Реббек К.А., Обендорф Д., Конлан С., Бахло М., Близзард К.А., Пайкрофт С., Крайсс А.
, Келлис М., Старк А., Харкинс Т.Т., Маршалл Graves JA, Woods GM, Hanon GJ, Papenfuss AT: Транскриптом тасманского дьявола раскрывает происхождение шванновских клеток клонально передающегося рака. Наука. 2010, 327 (5961): 84-87. 10.1126/научн.1180616.КАС Статья Google ученый
Парчман Т.Л., Гейст К.С., Гранен Дж.А., Бенкман К.В., Бюркле К.А.: Секвенирование транскриптомов экологически важных видов деревьев: сборка, аннотация и обнаружение маркеров. Геномика BMC. 2010, 11:
Google ученый
Бабик В., Стуглик М., Ци В., Куенцли М., Кудук К., Котеджа П., Радван Дж.: Транскриптом сердца рыжей полевки (Myodes glareolus): к пониманию эволюционных изменений скорости метаболизма. Геномика BMC. 2010, 11:390-10.1186/1471-2164-11-390.
Артикул Google ученый
Williams CE, Stclair DA: Фенетические взаимосвязи и уровни изменчивости, обнаруженные с помощью полиморфизма длины рестрикционных фрагментов и анализа случайной амплификации полиморфной ДНК культивируемых и диких образцов Lycopersicon-esculentum. Геном. 1993, 36 (3): 619-630. 10.1139/г93-083.
КАС Статья Google ученый
Рик CM: Томат, Lycopersicon esculentum (Solanaceae). Эволюция культурных растений. Под редакцией: Симмондс Н.В. 1976, Лондон: Longman Group, 268–273.
Google ученый
Лабате Дж. А., Бальдо А. М.: Обнаружение SNP помидоров с помощью EST-анализа и повторного секвенирования.
Молекулярная селекция. 2005, 16 (4): 343-349. 10.1007/s11032-005-1911-5.КАС Статья Google ученый
Yano K, Watanabe M, Yamamoto N, Maeda F, Tsugane T, Shibata D: База данных меток экспрессированной последовательности (EST) миниатюрного сорта томата, Micro-Tom. Физиология растений и клеток. 2005, 46: С139-С139.
Google ученый
Хименес-Гомез Дж. М., Малуф Дж. Н.: Разнообразие последовательностей у трех видов томатов: SNP, маркеры и молекулярная эволюция. BMC Биология растений. 2009 г., 9:
Google ученый
Yang WC, Bai XD, Kabelka E, Eaton C, Kamoun S, van der Knaap E, Francis D: Открытие полиморфизма одиночных нуклеотидов в Lycopersicon esculentum с помощью компьютерного анализа выраженных тегов последовательности. Молекулярная селекция.
2004, 14 (1): 21-34.КАС Статья Google ученый
Ван Дейнзе А., Стоффель К., Бьюэлл К.Р., Козик А., Лю Дж., Ван дер Кнаап Э., Фрэнсис Д.: Разнообразие консервативных генов томата. Геномика BMC. 2007, 8:
Google ученый
Сим С.К., Роббинс М.Д., Чилкотт С., Чжу Т., Фрэнсис Д.М.: Открытие массива олигонуклеотидов полиморфизмов в культивируемом томате (Solanum lycopersicum L.) выявило закономерности вариаций SNP, связанных с размножением. Геномика BMC. 2009, 10:
Google ученый
Биоинформатика в COMAV. [http://bioinf.comav.upv.es/ngs_backbone/index.html]
Широкий институт. [http://www.broadinstitute.org/igv]
Биоинформатика в COMAV. [http://bioinf.comav.upv.es/ngs_backbone/install.
html]Социальное кодирование Github. [http://github.com/JoseBlanca/franklin]
Чоу Х.Х., Холмс М.Х.: Качественная обрезка последовательности ДНК и удаление вектора. Биоинформатика. 2001, 17 (12): 1093-1104. 10.1093/биоинформатика/17.12.1093.
КАС Статья Google ученый
Пикард. [http://picard.sourceforge.net/index.shtml]
Маккенна А., Ханна М., Бэнкс Э., Сиваченко А., Цитулскис К., Керницкий А., Гаримелла К., Альтшулер Д., Габриэль С., Дали М., ДеПристо MA: Набор инструментов для анализа генома: платформа MapReduce для анализа данных секвенирования ДНК следующего поколения. Геномные исследования. 2010, 20: 1297-1303. 10.1101/гр.107524.110.
КАС Статья Google ученый
Солнечная геномная сеть. [ftp://ftp.
solgenomics.net/]NCBI Генбанк. [http://www.ncbi.nlm.nih.gov/genbank/]
Gundry CN, Vandersteen JG, Reed GH, Pryor RJ, Chen J, Wittwer CT: Анализ плавления ампликона с мечеными праймерами: пробирочный метод для дифференциации гомозигот и гетерозигот. Клиническая химия. 2003, 49 (3): 396-406. 10.1373/49.3.396.
КАС Статья Google ученый
 Геномные исследования. 2004, 14 (6): 1147-1159. 10.1101/гр.1917404.
Геномные исследования. 2004, 14 (6): 1147-1159. 10.1101/гр.1917404. 10.1093/биоинформатика/btp352.
10.1093/биоинформатика/btp352. Х., Хеккель Д.Г.: Транскриптомы следующего поколения для геномов следующего поколения с использованием сборки est2. БМК Биоинформатика. 2009, 10:
Х., Хеккель Д.Г.: Транскриптомы следующего поколения для геномов следующего поколения с использованием сборки est2. БМК Биоинформатика. 2009, 10: , Келлис М., Старк А., Харкинс Т.Т., Маршалл Graves JA, Woods GM, Hanon GJ, Papenfuss AT: Транскриптом тасманского дьявола раскрывает происхождение шванновских клеток клонально передающегося рака. Наука. 2010, 327 (5961): 84-87. 10.1126/научн.1180616.
, Келлис М., Старк А., Харкинс Т.Т., Маршалл Graves JA, Woods GM, Hanon GJ, Papenfuss AT: Транскриптом тасманского дьявола раскрывает происхождение шванновских клеток клонально передающегося рака. Наука. 2010, 327 (5961): 84-87. 10.1126/научн.1180616. “>
“>Миллер Дж. К., Танксли С. Д.: RFLP-анализ филогенетических отношений и генетической изменчивости в роде Lycopersicon. Теоретическая и прикладная генетика. 1990, 80 (4): 437-448.
КАС Статья Google ученый
 Молекулярная селекция. 2005, 16 (4): 343-349. 10.1007/s11032-005-1911-5.
Молекулярная селекция. 2005, 16 (4): 343-349. 10.1007/s11032-005-1911-5. 2004, 14 (1): 21-34.
2004, 14 (1): 21-34.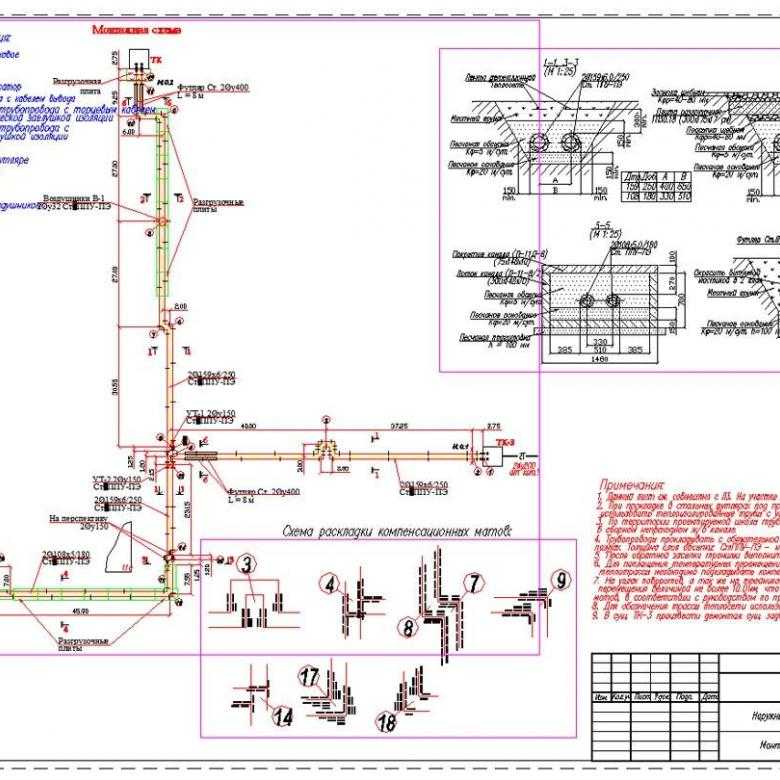 html]
html] solgenomics.net/]
solgenomics.net/]Скачать ссылки
Благодарности
Авторы благодарят COMAV за любезно предоставленные образцы томатов. Мы также выражаем благодарность Джошуа Бергену за помощь в улучшении английского языка этой рукописи.
Информация об авторе
Примечания автора
Авторы и организации
Instituto de Conservación y Mejora de la Agrodiversidad Valenciana (COMAV)0041
Jose M Blanca, Laura Pascual, Peio Ziarsolo, Fernando Nuez и Joaquin Cañizares
Авторы
- Jose M Blanca
Посмотреть публикации автора
Вы также можете искать этого автора в PubMed Google Scholar
- Laura Pascual
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
- Peio Ziarsolo
Посмотреть публикации автора
Вы также можете искать этого автора в PubMed Google Scholar
- Fernando Nuez
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
- Joaquin Cañizares
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
Автор, ответственный за корреспонденцию
Хоакин Каньисарес.
Дополнительная информация
Вклад авторов
LP получила экспериментальные данные и участвовала в анализе. JC разработал исследование и эксперименты и участвовал в анализе. JMB и PZ разработали программное обеспечение ngs-backbone и участвовали в анализе. FN отбирала и обрабатывала растительный материал. JMB, LP, FN и JC написали рукопись. Все авторы прочитали и одобрили окончательный вариант рукописи.
Хосе М. Бланка, Лаура Паскуаль внесли одинаковый вклад в эту работу.
Электронный дополнительный материал
Дополнительный файл 1:ngs_backbone 1.1.0 software. ngs_backbone 1.1.0. Последняя версия, выпущенная 31 августа 2010 г. (GZ 19 MB)
Доп. файл 2:Материалы и методы. материалы и методы для валидации экспериментального программного обеспечения. (PDF, 87 КБ)
12864_2010_10168_MOESM3_ESM.VCF
Дополнительный файл 3: файл данных VCF, содержащий все идентифицированные SNV. Файл данных, содержащий все идентифицированные SNV. SNV не были выбраны с каким-либо дополнительным фильтром, но файл включает данные для всех фильтров ngs_backbone, используемых в этой работе. (ВКФ 17 МБ)
SNV не были выбраны с каким-либо дополнительным фильтром, но файл включает данные для всех фильтров ngs_backbone, используемых в этой работе. (ВКФ 17 МБ)
12864_2010_10168_MOESM4_ESM.CSV
Дополнительный файл 4: Доступы, используемые при проверке SNV. Староместные сорта были предоставлены генбанком COMAV, коммерческие сорта получены от Semilleros Cucala Agricola (Beniganim, Испания). (CSV 1000 байт)
Дополнительный файл 5:Последовательности праймеров. Файл с последовательностями праймеров, используемых в данной работе. (CSV 5 КБ)
Исходные файлы изображений, представленные авторами
Ниже приведены ссылки на исходные файлы изображений, представленные авторами.
Исходный файл авторов для рисунка 1
Права и разрешения
Эта статья находится под лицензией Creative Commons Attribution 4.0 International License, которая разрешает использование, совместное использование, адаптацию, распространение и воспроизведение на любом носителе или в любом формате при условии по мере того, как вы укажете первоначальных авторов и источник, предоставьте ссылку на лицензию Creative Commons и укажите, были ли внесены изменения. Изображения или другие сторонние материалы в этой статье включены в лицензию Creative Commons на статью, если иное не указано в кредитной строке материала. Если материал не включен в лицензию Creative Commons статьи, а ваше предполагаемое использование не разрешено законом или выходит за рамки разрешенного использования, вам необходимо получить разрешение непосредственно от правообладателя. Чтобы просмотреть копию этой лицензии, посетите http://creativecommons.org/licenses/by/4.0/. Отказ Creative Commons от права на общественное достояние (http://creativecommons.org/publicdomain/zero/1.0/) применяется к данным, представленным в этой статье, если иное не указано в кредитной линии данных.
Изображения или другие сторонние материалы в этой статье включены в лицензию Creative Commons на статью, если иное не указано в кредитной строке материала. Если материал не включен в лицензию Creative Commons статьи, а ваше предполагаемое использование не разрешено законом или выходит за рамки разрешенного использования, вам необходимо получить разрешение непосредственно от правообладателя. Чтобы просмотреть копию этой лицензии, посетите http://creativecommons.org/licenses/by/4.0/. Отказ Creative Commons от права на общественное достояние (http://creativecommons.org/publicdomain/zero/1.0/) применяется к данным, представленным в этой статье, если иное не указано в кредитной линии данных.
Перепечатки и разрешения
Об этой статье
Fast-GBS: новый конвейер для эффективного и высокоточного определения SNP на основе данных генотипирования путем секвенирования | BMC Биоинформатика
- Программное обеспечение
- Открытый доступ
- Опубликовано:
- Давуд Торкамане 1,2 ,
- Jérôme Laroche 2 ,
- Maxime Bastien 1,2 ,
- Amina Abed 1,2 &
- …
- François Belzile 1,2
Биоинформатика BMC том 18 , номер статьи: 5 (2017) Процитировать эту статью
11 тыс. обращений
67 цитирований
3 Альтметрический
Сведения о показателях
Abstract
Background
Технологии секвенирования следующего поколения (NGS) значительно ускорили изучение состава геномов и их функций. Генотипирование посредством секвенирования (GBS) — это подход к генотипированию, который использует NGS для быстрого и экономичного сканирования генома. Было показано, что он позволяет одновременно обнаруживать и генотипировать от тысяч до миллионов SNP у широкого круга видов. Для большинства пользователей основной проблемой GBS является биоинформатический анализ большого количества информации о последовательностях, полученной в результате секвенирования библиотек GBS, с точки зрения вызова аллелей в локусах SNP. Здесь мы описываем новый конвейер биоинформатики GBS, Fast-GBS, предназначенный для обеспечения высокоточного генотипирования, требующий скромных вычислительных ресурсов и обеспечивающий простоту использования.
Генотипирование посредством секвенирования (GBS) — это подход к генотипированию, который использует NGS для быстрого и экономичного сканирования генома. Было показано, что он позволяет одновременно обнаруживать и генотипировать от тысяч до миллионов SNP у широкого круга видов. Для большинства пользователей основной проблемой GBS является биоинформатический анализ большого количества информации о последовательностях, полученной в результате секвенирования библиотек GBS, с точки зрения вызова аллелей в локусах SNP. Здесь мы описываем новый конвейер биоинформатики GBS, Fast-GBS, предназначенный для обеспечения высокоточного генотипирования, требующий скромных вычислительных ресурсов и обеспечивающий простоту использования.
Результаты
Fast-GBS построен на стандартном языке биоинформатики и форматах файлов, способен обрабатывать данные с разных платформ секвенирования, способен обнаруживать различные виды вариантов (SNP, MNP и Indels). Чтобы проиллюстрировать его эффективность, мы назвали варианты в трех коллекциях образцов (сои, ячменя и картофеля), которые охватывают диапазон различных размеров генома, уровней сложности генома и плоидности. В рамках этих небольших наборов образцов мы назвали 35 тыс., 32 тыс. и 38 тыс. SNP для сои, ячменя и картофеля соответственно. Чтобы оценить точность генотипа, мы сравнили эти генотипы SNP, полученные из GBS, с независимыми наборами данных, полученными в результате полногеномного секвенирования или массивов SNP. Этот анализ дал оценочную точность 98,7, 95,2 и 94% для сои, ячменя и картофеля соответственно.
В рамках этих небольших наборов образцов мы назвали 35 тыс., 32 тыс. и 38 тыс. SNP для сои, ячменя и картофеля соответственно. Чтобы оценить точность генотипа, мы сравнили эти генотипы SNP, полученные из GBS, с независимыми наборами данных, полученными в результате полногеномного секвенирования или массивов SNP. Этот анализ дал оценочную точность 98,7, 95,2 и 94% для сои, ячменя и картофеля соответственно.
Выводы
Мы пришли к выводу, что Fast-GBS предоставляет высокоэффективный и надежный инструмент для вызова SNP из данных GBS.
История вопроса
В настоящее время геномика лежит в основе невероятного количества открытий, инноваций и приложений. Эта революция является прямым результатом появления технологий секвенирования следующего поколения (NGS) [1–4]. В области генотипирования сочетание NGS и методов редуцированного представления, которые фокусируют усилия по секвенированию на небольшом подмножестве генома, сделало возможным одновременное обнаружение однонуклеотидного полиморфизма (SNP) и генотипирование всего генома в одном месте. шаг даже у видов с большими геномами [5]. Это значительно облегчило генотипирование очень большого количества SNP с использованием ряда родственных методов (например, CRoPS, RAD-seq, GBS, RAD-seq с двойным перевариванием и 2bRAD) [6–11]. Эти разнообразные методы позволяют изучать важные вопросы молекулярной селекции, популяционной генетики, экологической генетики и эволюции с использованием от тысяч до миллионов генетических маркеров у широкого круга видов [5]. Генотипирование путем секвенирования (GBS) является особенно привлекательным методом снижения сложности, который предлагает простой, надежный, недорогой и высокопроизводительный метод генотипирования как модельных, так и немодельных видов [8].
шаг даже у видов с большими геномами [5]. Это значительно облегчило генотипирование очень большого количества SNP с использованием ряда родственных методов (например, CRoPS, RAD-seq, GBS, RAD-seq с двойным перевариванием и 2bRAD) [6–11]. Эти разнообразные методы позволяют изучать важные вопросы молекулярной селекции, популяционной генетики, экологической генетики и эволюции с использованием от тысяч до миллионов генетических маркеров у широкого круга видов [5]. Генотипирование путем секвенирования (GBS) является особенно привлекательным методом снижения сложности, который предлагает простой, надежный, недорогой и высокопроизводительный метод генотипирования как модельных, так и немодельных видов [8].
Усовершенствованные технологии секвенирования (NGS) сократили как стоимость, так и время, необходимое для создания данных секвенирования. Эффективная и точная вычислительная обработка, определение вариантов и генотипов крупномасштабных данных NGS является новым узким местом в геномике. Чтобы удовлетворить эту потребность, были разработаны многочисленные конвейеры биоинформатики [12–16], и все они должны выполнять аналогичный набор шагов, таких как: 1) получение необработанных данных о последовательности, 2) демультиплексирование объединенных данных чтения последовательности, 3) фильтрация низкоуровневых данных. качественные риды, 4) сборка или выравнивание ридов и, наконец, 5) обнаружение полиморфных локусов и вывод о реальных генотипах в этих локусах. Каждый шаг представляет собой набор конкретных проблем и неясностей. Как правило, этим проблемам могут способствовать различные геномные характеристики, такие как количество обнаруженных вариантов, сложность генома, степень гетерозиготности, доля повторяющихся последовательностей во всем геноме, уровень полиморфизма и дивергенция среди популяций [12]. . С другой стороны, технические факторы, такие как качество ДНК, степень мультиплексирования образца, общее количество прочтений на образец, длина прочтений и частота ошибок секвенирования, взаимодействуют с этими биологическими факторами [17–19].
Чтобы удовлетворить эту потребность, были разработаны многочисленные конвейеры биоинформатики [12–16], и все они должны выполнять аналогичный набор шагов, таких как: 1) получение необработанных данных о последовательности, 2) демультиплексирование объединенных данных чтения последовательности, 3) фильтрация низкоуровневых данных. качественные риды, 4) сборка или выравнивание ридов и, наконец, 5) обнаружение полиморфных локусов и вывод о реальных генотипах в этих локусах. Каждый шаг представляет собой набор конкретных проблем и неясностей. Как правило, этим проблемам могут способствовать различные геномные характеристики, такие как количество обнаруженных вариантов, сложность генома, степень гетерозиготности, доля повторяющихся последовательностей во всем геноме, уровень полиморфизма и дивергенция среди популяций [12]. . С другой стороны, технические факторы, такие как качество ДНК, степень мультиплексирования образца, общее количество прочтений на образец, длина прочтений и частота ошибок секвенирования, взаимодействуют с этими биологическими факторами [17–19]. ]. Следовательно, необходимо выбрать соответствующие параметры, такие как требуемая глубина охвата, качество картирования чтения или допустимая степень расхождения для успешного картирования. Наконец, из-за этих двух разных источников изменчивости (биологический и технический) в данных GBS оптимальные параметры должны быть скорректированы для каждого вида и желаемого охвата и пропускной способности маркеров.
]. Следовательно, необходимо выбрать соответствующие параметры, такие как требуемая глубина охвата, качество картирования чтения или допустимая степень расхождения для успешного картирования. Наконец, из-за этих двух разных источников изменчивости (биологический и технический) в данных GBS оптимальные параметры должны быть скорректированы для каждого вида и желаемого охвата и пропускной способности маркеров.
Обычно биоинформатические конвейеры для обработки данных GBS подразделяются на две группы: de novo на основе и на основе ссылок. При наличии эталонного генома считывания секвенирования с уменьшенным представлением могут быть сопоставлены с эталонным геномом и могут быть вызваны SNP [12, 20]. К настоящему времени было разработано несколько конвейеров анализа GBS на основе эталонов. Наиболее широко используемыми конвейерами анализа GBS на основе эталонов являются: TASSEL-GBS (v1 и v2), Stacks и IGST [13–15, 21]. Но когда эталонный геном недоступен, необходимо идентифицировать пары почти идентичных прочтений (предположительно представляющих альтернативные аллели в локусе). Наиболее часто используемые конвейеры для такого de novo — UNEAK и Stacks [15, 16].
Наиболее часто используемые конвейеры для такого de novo — UNEAK и Stacks [15, 16].
Здесь мы описываем новый эталонный пайплайн Fast-GBS и оцениваем пайплайн на основе крупномасштабного анализа видов сои, ячменя и картофеля. Его легко использовать с разными видами в разных контекстах, и он предоставляет аналитическую платформу, которую можно запускать с различными типами данных секвенирования и скромными вычислительными ресурсами.
Тестовые наборы данных
Для проверки производительности Fast-GBS мы использовали существующие наборы данных последовательностей для панелей из 24 неродственных образцов/клонов для трех видов, охватывающих ряд геномных ситуаций: соя [22], ячмень [Abed et al., неопубликовано] и картофеля [Bastien et al., неопубликовано]. В таблице 1 показаны виды, которые мы использовали в этом исследовании. Они различаются по своей плоидности, размеру генома и способу воспроизводства (что связано с ожидаемой зиготностью). Мы использовали наборы данных последовательностей, состоящие из 24 образцов для каждого вида.
 SNP) [23].
SNP) [23].Внедрение
Конвейер анализа Fast-GBS был разработан путем интеграции общедоступных пакетов с инструментами собственной разработки. Общедоступные пакеты включают Sabre (демультиплексирование) [24], Cutadapt (обрезка и очистка чтения) [25], BWA (отображение чтения) [26], SAMtools (преобразование и индексирование файлов) [27] и Platypus (постобработка файлов). риды, построение гаплотипов и определение вариантов) [28]. Функции и программные средства Fast-GBS представлены на рис. 1.
рис. 1Схематическое представление аналитических шагов в конвейере Fast-GBS. Основные этапы аналитического процесса указаны в центральной части диаграммы, а различные используемые программные средства указаны слева, а входы и выходы каждого шага — справа
Полноразмерное изображение
Создание структуры каталогов
Мы разработали сценарий Bash для создания структуры каталогов перед запуском конвейера Fast-GBS. Эта командная строка создает каталоги для данных (файлы FASTQ), штрих-коды (ключевой файл), эталонный геном и результаты (выходные данные Fast-GBS).
Входные данные
Входные данные представляют собой секвенированные фрагменты ДНК из любого протокола GBS на основе рестрикционных ферментов. Fast-GBS обрабатывает необработанные данные секвенирования в формате FASTQ.
Подготовка файла параметров
Файл параметров представляет собой текстовый файл, содержащий ключевую информацию об анализе, включая путь к файлам FASTQ, штрих-коды и эталонный геном. Он также содержит информацию о типе последовательности (парная или односторонняя), последовательности адаптера и технологии секвенирования. В этом файле мы можем определить критические параметры фильтрации, такие как минимальные показатели качества для прочтений, минимальное количество прочтений, необходимых для определения генотипа, и максимально допустимое количество отсутствующих данных. В этом файле также определяется количество ЦП, имена выходных файлов. Этот файл поставляется с конвейером Fast-GBS.
Демультиплексирование данных
Экономическая эффективность GBS частично обусловлена мультиплексированием выборок, и результирующие объединенные чтения должны быть демультиплексированы перед вызовом SNP. Fast-GBS использует Sabre [24] для демультиплексирования считываний штрих-кодов в отдельные файлы. Он просто сравнивает предоставленные штрих-коды с 5′-концом каждого считывания и разделяет считывания на соответствующие файлы штрих-кодов после вырезания штрих-кода из считывания. Если считывание не имеет распознанного штрих-кода, оно помещается в «неизвестный» файл. В Sabre также есть опция (-m), разрешающая несовпадения штрих-кодов. Sabre поддерживает входные файлы, сжатые gzip. После демультиплексирования Sabre выводит файл журнала сводки BC о том, сколько считываний было выполнено в каждом файле штрих-кода.
Fast-GBS использует Sabre [24] для демультиплексирования считываний штрих-кодов в отдельные файлы. Он просто сравнивает предоставленные штрих-коды с 5′-концом каждого считывания и разделяет считывания на соответствующие файлы штрих-кодов после вырезания штрих-кода из считывания. Если считывание не имеет распознанного штрих-кода, оно помещается в «неизвестный» файл. В Sabre также есть опция (-m), разрешающая несовпадения штрих-кодов. Sabre поддерживает входные файлы, сжатые gzip. После демультиплексирования Sabre выводит файл журнала сводки BC о том, сколько считываний было выполнено в каждом файле штрих-кода.
Обрезка и очистка
После демультиплексирования Fast-GBS использует Cutadapt для поиска и удаления последовательностей адаптеров, праймеров и других типов нежелательных последовательностей из считываний высокопроизводительного секвенирования [25].
Алгоритмы сопоставления чтения
Fast-GBS использует алгоритм MEM (максимальные точные совпадения), реализованный в BWA, который работает путем заполнения выравниваний и последующего расширения начальных значений с помощью алгоритма Смита-Уотермана (SW) с использованием штрафа за аффинный пробел [26]. Этот алгоритм может выполнять локальное выравнивание для операций чтения от 70 до 1 Мбит/с. Этот алгоритм может выполнять параллельное выравнивание, что заметно увеличивает скорость анализа. Возможность выравнивания прочтений переменного размера позволяет использовать данные, полученные с помощью разных платформ секвенирования (Illumina, Ion Torrent и др.). Выровненные чтения могут быть разделены промежутками, чтобы учесть вставки.
Этот алгоритм может выполнять локальное выравнивание для операций чтения от 70 до 1 Мбит/с. Этот алгоритм может выполнять параллельное выравнивание, что заметно увеличивает скорость анализа. Возможность выравнивания прочтений переменного размера позволяет использовать данные, полученные с помощью разных платформ секвенирования (Illumina, Ion Torrent и др.). Выровненные чтения могут быть разделены промежутками, чтобы учесть вставки.
Постобработка сопоставленных прочтений
После начального выравнивания сопоставленные чтения дополнительно обрабатываются Platypus [28] для улучшения чувствительности и специфичности определения вариантов. Эта постобработка направлена на улучшение качества сопоставления путем повторного изучения плохо сопоставленных прочтений и сопоставления прочтений с несколькими местоположениями. Platypus классифицирует плохо картированные чтения по трем категориям: 1) чтения с многочисленными несовпадениями (высокий уровень ошибок секвенирования), 2) чтения, сопоставленные с несколькими участками генома, и 3) любые оставшиеся последовательности линкера или адаптера (вызывающие плохое картирование). Варианты, вызванные с использованием таких потенциально неправильно сопоставленных чтений (см. следующий шаг), выделяются с помощью флага BadReads.
Варианты, вызванные с использованием таких потенциально неправильно сопоставленных чтений (см. следующий шаг), выделяются с помощью флага BadReads.
Построение гаплотипов и вызов вариантов
В Fast-GBS варианты вызываются с использованием Platypus. В отличие от вызывающих вариантов на основе выравнивания, которые фокусируются на одном типе варианта (SNP или indel), Platypus использует вызов вариантов с несколькими образцами, который помогает использовать информацию из нескольких образцов для вызова вариантов, которые могут выглядеть ненадежными в одном образце. Этот подход уменьшает количество ошибок, связанных с вставками и более крупными вариантами (MNP). Сначала локальный ассемблер просматривает небольшое окно (~несколько килобайт) за раз и использует все чтения в окне для создания цветного графа де Брейна, затем, используя все варианты-кандидаты, он генерирует исчерпывающий список гаплотипов. Гаплотипы-кандидаты генерируются путем кластеризации аллелей-кандидатов по окнам. Частоты гаплотипов оцениваются с помощью алгоритма максимизации ожидания (EM). Затем варианты называют с использованием предполагаемых частот гаплотипов. Этот подход работает на уровне локальных гаплотипов, а не на уровне отдельных вариантов, и хорошо работает в сильно различающихся регионах. Это также снижает требования к вычислительным ресурсам.
Частоты гаплотипов оцениваются с помощью алгоритма максимизации ожидания (EM). Затем варианты называют с использованием предполагаемых частот гаплотипов. Этот подход работает на уровне локальных гаплотипов, а не на уровне отдельных вариантов, и хорошо работает в сильно различающихся регионах. Это также снижает требования к вычислительным ресурсам.
Фильтрация вариантов и индивидуумов
Platypus изначально был разработан и использовался для обнаружения вариантов в образцах человека, мыши, крысы и шимпанзе. Чтобы оптимизировать параметры Platypus в контексте анализа односторонних чтений, полученных из GBS, мы изменили несколько параметров (см. [29] для получения подробной информации о параметрах Platypus). Некоторые из фильтров, используемых на этапах вызова вариантов Fast-GBS: количество прочтений (NR) на локус (по умолчанию = 2), оценка качества сопоставления прочтений с вызовом варианта (MQ ≥ 10), минимальное базовое качество (по умолчанию = 10). ), расстояние MNP (minFlank = 5) и максимально допустимое количество отсутствующих данных (MaxMD) (по умолчанию ≤ 80%). Полное описание всех параметров фильтрации см. в руководстве пользователя Fast-GBS.
Полное описание всех параметров фильтрации см. в руководстве пользователя Fast-GBS.
Выходные данные
Основным выходным файлом Fast-GBS является файл .vcf [30], содержащий подробную информацию о каждом из вариантов. Кроме того, Fast-GBS также создает простой текстовый файл, содержащий только генотипические данные. Файл журнала Fast-GBS содержит завершенные шаги конвейера во время его работы. В случаях, когда возникает ошибка и преждевременно прекращается работа конвейера, в лог-файле отображается шаг, на котором остановился анализ. Анализ можно начать в любой точке существующих промежуточных файлов, просто создав файл журнала, в котором перечислены ранее выполненные шаги. Fast-GBS повторно инициирует анализ, начиная с этого момента.
Результаты и обсуждение
Производительность Fast-GBS
Чтобы оценить производительность конвейера анализа Fast-GBS, мы использовали его для анализа существующих данных чтения, полученных с помощью GBS, из наборов из 24 образцов сои, ячменя и картофеля. В таблице 2 представлены сводные данные этого анализа. Как можно видеть, в общей сложности было вызвано 35 тыс. SNP с использованием 42 M чтений Illumina размером 100 п.н. на ДНК, расщепленной Ape KI, из 24 различных линий сои. Аналогичным образом, для ячменя 32 тыс. SNP были успешно вызваны из 72 млн прочтений Ion Torrent (длиной 50–150 п.н.), полученных из 24-плекса Msp I/ Pst I библиотека. Наконец, у картофеля 38 тыс. SNP были получены в результате секвенирования 24-плексной библиотеки Msp I/ Pst I (43 миллиона прочтений Illumina по 100 п.н.).
В таблице 2 представлены сводные данные этого анализа. Как можно видеть, в общей сложности было вызвано 35 тыс. SNP с использованием 42 M чтений Illumina размером 100 п.н. на ДНК, расщепленной Ape KI, из 24 различных линий сои. Аналогичным образом, для ячменя 32 тыс. SNP были успешно вызваны из 72 млн прочтений Ion Torrent (длиной 50–150 п.н.), полученных из 24-плекса Msp I/ Pst I библиотека. Наконец, у картофеля 38 тыс. SNP были получены в результате секвенирования 24-плексной библиотеки Msp I/ Pst I (43 миллиона прочтений Illumina по 100 п.н.).
 Анализ этого набора данных с использованием IGST занял четыре дня, в то время как тот же анализ с использованием Fast-GBS при том же размере выборки и условиях занял всего 11 часов и вызвал ~ 60 тыс. SNP (данные не показаны). Как видно, Fast-GBS обеспечивает высокий уровень производительности для образцов сои.
Анализ этого набора данных с использованием IGST занял четыре дня, в то время как тот же анализ с использованием Fast-GBS при том же размере выборки и условиях занял всего 11 часов и вызвал ~ 60 тыс. SNP (данные не показаны). Как видно, Fast-GBS обеспечивает высокий уровень производительности для образцов сои. Ячмень имеет один из самых больших геномов (>5 Гб) среди культурных растений. Из-за огромного размера и высокого уровня сложности его генома настоятельно рекомендуется снижение сложности ячменя, важного вида сельскохозяйственных культур, для которого был опубликован проект генома [32]. Mascher et al [33] генотипировали 94 линии RIL ячменя, используя GBS (библиотека Msp I/ Pst I) и назвали 34 тыс. и 19 тыс. SNP, используя либо эталонный геном (с помощью SAMtools), либо de novo. 9трубопровод 0038 (ТАССЕЛЬ) соответственно. В этом исследовании мы использовали Fast-GBS для вызова SNP в ячмене, и, как видно из таблицы 2, Fast-GBS вызвал 32 тыс. SNP для небольшого количества образцов (24). Это показало способность Fast-GBS работать с большими и сложными геномами.
Это показало способность Fast-GBS работать с большими и сложными геномами.
Из-за высокого уровня плоидности и гетерозиготности картофель является сложным видом для генотипирования. Наиболее часто используемым методом генотипирования картофеля является массив SNP. К настоящему времени разработаны два массива SNP: массивы SolCAP 8k и 20k [23, 34, 35]. Недавно Endelman [36] генотипировал 96 образцов диплоидного картофеля F2 с использованием GBS. Используя конвейер биоинформатики на основе R для фильтрации вариантов GBS, они идентифицировали 11 тыс. SNP. В этом исследовании мы назвали 38 тыс. SNP из 24 образцов, которые также были генотипированы с использованием матрицы SolCAP 8 тыс. SNP. Используя упрощенный режим генотипирования («диплоидный режим»), в котором различались только три генотипических класса (0/0, 0/1 и 1/1), было обнаружено, что 5,5 тыс. SNP на этом массиве полиморфны среди этого набора из 24. образцы картофеля [37]. Как видно из таблицы 2, при использовании Fast-GBS для вызова SNP в эквивалентном диплоидном режиме мы вызвали почти в семь раз больше полиморфизмов, чем при использовании массива SNP (38 тыс.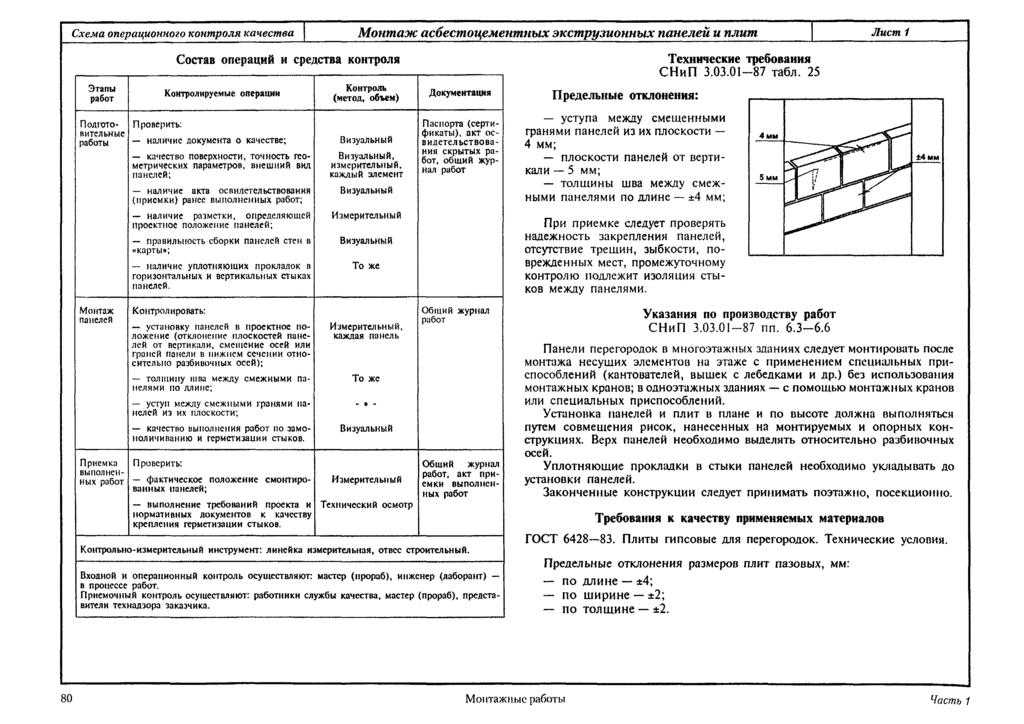 против 5,5 тыс. SNP).
против 5,5 тыс. SNP).
Проверка данных Fast-GBS
Важным аспектом, который следует учитывать при использовании любого инструмента для определения вариантов, является точность вызываемых генотипов. В этом исследовании мы оценили точность генотипов, названных Fast-GBS (таблица 2), сравнив их с «истинными» генотипами (полученными либо с помощью полногеномного повторного секвенирования, либо с помощью данных массива SNP). Для сои для всех 24 образцов мы сравнили генотипы SNP, названные Fast-GBS, с генотипами, назначенными тем же локусам после полногеномного секвенирования. Мы обнаружили очень высокий уровень конкордантности, так как почти все генотипы (98,7%) оказались идентичными. Для ячменя мы сравнили генотипы SNP, названные Fast-GBS, с истинными генотипами для одной из 24 линий (сорт Morex), единственной, для которой у нас были данные полногеномного секвенирования. Опять же, была получена высокая степень согласия между двумя наборами данных (97%). Наконец, для картофеля мы использовали данные, полученные на чипе SolCAP 8k Infinium для тех же 24 образцов, которые использовались для выполнения GBS. Эти два набора данных имеют общие 122 локуса SNP. В нашем первоначальном сравнении только 87,7% были согласны. Когда мы изучили пропорцию конкордантных криков, мы обнаружили, что более 50% всех несогласных криков исходили только от трех образцов, и степень дискордантности в них была настолько велика, что предполагалось, что мы не сравниваем одни и те же клоны. После удаления этих выбросов из анализа 94% генотипов, названных Fast-GBS и массивом SNP, совпадали в оставшихся 21 клоне. Мы пришли к выводу, что Fast-GBS может точно вызывать SNP у видов с разными характеристиками (размер генома, плоидность, зиготность).
Эти два набора данных имеют общие 122 локуса SNP. В нашем первоначальном сравнении только 87,7% были согласны. Когда мы изучили пропорцию конкордантных криков, мы обнаружили, что более 50% всех несогласных криков исходили только от трех образцов, и степень дискордантности в них была настолько велика, что предполагалось, что мы не сравниваем одни и те же клоны. После удаления этих выбросов из анализа 94% генотипов, названных Fast-GBS и массивом SNP, совпадали в оставшихся 21 клоне. Мы пришли к выводу, что Fast-GBS может точно вызывать SNP у видов с разными характеристиками (размер генома, плоидность, зиготность).
Гибкость для запуска различных платформ секвенирования
В этом исследовании для оценки производительности Fast-GBS мы использовали чтения Illumina и Ion Torrent. Образцы сои и картофеля секвенировали с использованием платформы Illumina Hiseq, а образцы ячменя — с помощью платформы Ion Torrent (Proton). Как правило, для GBS секвенирование Illumina генерирует считывания одинаковой длины (100 п. н.), в то время как считывания Ion Torrent составляют от 50 до 150 п.н. Секвенирование Ion Torrent обычно приводит к более высокому уровню ошибок секвенирования [38, 39].]. Таким образом, предпочтительно, чтобы аналитический конвейер был универсальным и мог использовать чтения, полученные с помощью любой технологии (или новых технологий в разработке). Большинство биоинформатических конвейеров GBS могут работать с чтением Ion Torrent, но их часто необходимо модифицировать, чтобы они подходили для этого типа данных чтения. TASSEL, UNEAK и Stacks генерируют теги фиксированной длины (например, 64 п.н.). Это приведет к важной потере информации о последовательности и может привести к неточному или неоднозначному отображению прочтений. Кроме того, из-за повышенного количества ошибок секвенирования эти конвейеры могут генерировать ложные теги, которые создают ложные SNP. Как показано выше, Fast-GBS доказал способность точно выполнять вызов максимального SNP с использованием ридов, полученных с обеих платформ секвенирования (Ion Proton и Illumina).
н.), в то время как считывания Ion Torrent составляют от 50 до 150 п.н. Секвенирование Ion Torrent обычно приводит к более высокому уровню ошибок секвенирования [38, 39].]. Таким образом, предпочтительно, чтобы аналитический конвейер был универсальным и мог использовать чтения, полученные с помощью любой технологии (или новых технологий в разработке). Большинство биоинформатических конвейеров GBS могут работать с чтением Ion Torrent, но их часто необходимо модифицировать, чтобы они подходили для этого типа данных чтения. TASSEL, UNEAK и Stacks генерируют теги фиксированной длины (например, 64 п.н.). Это приведет к важной потере информации о последовательности и может привести к неточному или неоднозначному отображению прочтений. Кроме того, из-за повышенного количества ошибок секвенирования эти конвейеры могут генерировать ложные теги, которые создают ложные SNP. Как показано выше, Fast-GBS доказал способность точно выполнять вызов максимального SNP с использованием ридов, полученных с обеих платформ секвенирования (Ion Proton и Illumina).
Выводы
GBS представляет собой чрезвычайно мощный и универсальный инструмент для идентификации и вызова генетических маркеров, который может использоваться исследователями, работающими с многочисленными видами и областями исследования. Этот подход к генотипированию, как и все приложения, основанные на NGS, генерирует огромное количество необработанных данных. Эти данные необходимо анализировать как можно быстрее и эффективнее, при этом получая данные SNP с высокой точностью. Fast-GBS зарекомендовал себя как мощный конвейер для создания большого количества высокоточных SNP с использованием данных чтения последовательностей, полученных с разных платформ секвенирования и различных видов, характеризующихся разными уровнями плоидности, зиготности и сложности генома. Сочетая таким образом эффективность и точность, Fast-GSB представляет собой полезный инструмент для широкого круга пользователей в различных исследовательских сообществах.
Наличие и требования
Название проекта: Fast-GBS
Домашняя страница проекта: https://bitbucket. org/jerlar73/fast-gbs
org/jerlar73/fast-gbs
Операционная система: Linux
Язык программирования: Bash и Python
Лицензия: GNU GPL v
Любые ограничения на использование неакадемическими работниками: Нет
Данные о последовательностях доступны в архиве NCBI Sequence Read Archive (SRA) с регистрационным номером SRP# Study, SRP059747.
Сокращения
- КУЛЬТУРЫ:
Снижение сложности полиморфных последовательностей
- ГБС:
Генотипирование путем секвенирования
- Индекс:
Вставка-удаление
- МаксМД:
Максимум отсутствующих данных
- МинМАФ:
Минимальная частота минорного аллеля
- минNR:
Минимальное количество операций чтения
- МНП:
Множественный полиморфизм нуклеотидов
- MQ:
Качество отображения
- NGS:
Секвенирование следующего поколения
- RAD-последовательность:
Секвенирование ДНК, связанное с сайтом рестрикции
- СНП:
Однонуклеотидный полиморфизм
Каталожные номера
Эдвардс Д., Бэтли Дж., Сноудон Р.Дж. Доступ к сложным геномам сельскохозяйственных культур с помощью секвенирования нового поколения. Теория Appl Genet. 2013; 126:1–11.
КАС Статья пабмед Google ученый
Килпинен Х., Барретт Дж.С. Как секвенирование нового поколения меняет генетику сложных заболеваний. Тенденции Жене. 2013;29:23–30.
КАС Статья пабмед Google ученый
Кумар С., Бэнкс Т.В., Клотье С. Открытие SNP с помощью секвенирования следующего поколения и его приложений. Int J Plant Genom. 2012. Дои: 10.1155/2012/831460.
Google ученый
Davey JW, Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML.
Открытие полногеномных генетических маркеров и генотипирование с использованием секвенирования нового поколения. Природа. 2011. doi: 10.1038/nrg3012.ПабМед Google ученый
van Orsouw NJ, Hogers RCJ, Janssen A, Yalcin F, Snoeijers S, Verstege E, et al. Снижение сложности полиморфных последовательностей (CRoPS): новый подход к обнаружению крупномасштабного полиморфизма в сложных геномах. ПЛОС Один. 2007;2, e1172.
Артикул пабмед ПабМед Центральный Google ученый
Эттер П.Д., Этвуд Т.С., Карри М.С., Шивер А.Л., Льюис З.А. и др. Быстрое обнаружение SNP и генетическое картирование с использованием секвенированных маркеров RAD. ПЛОС Один. 2007;3(10), e3376. doi:10.1371/journal.pone.0003376.
Google ученый
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al.
Надежный и простой подход генотипирования путем секвенирования (GBS) для видов с большим разнообразием. ПЛОС Один. 2011;6(5):19379.Артикул Google ученый
Эттер П.Д., Бэсшем С., Гогенлоэ П.А., Джонсон Э.А., Креско В.А. Открытие SNP и генотипирование для эволюционной генетики с использованием секвенирования RAD. Методы Мол Биол. 2001; 772: 157–78.
Артикул Google ученый
Петерсон Б.К., Вебер Дж.Н., Кей Э.Х., Фишер Х.С., Хекстра Х.Э. Двойной дайджест radseq: недорогой метод обнаружения SNP de novo и генотипирования у модельных и немодельных видов. ПЛОС Один. 2012;7, e37135.
КАС Статья пабмед ПабМед Центральный Google ученый
Ван С., Мейер Э., Маккей Дж. К., Мац М. В. 2b-RAD: простой и гибкий метод полногеномного генотипирования.
Нат Методы. 2012; 9: 808–10.КАС Статья пабмед Google ученый
Нильсен Р., Пол Дж. С., Альбрехтсен А., Сонг Ю. С. Вызов генотипа и SNP из данных секвенирования следующего поколения. Природа Преподобный Жене. 2011; 12:443–51.
КАС Статья пабмед ПабМед Центральный Google ученый
Брэдбери П.Дж., Чжан З., Крун Д.Э., Касстевенс Т.М., Рамдосс Ю., Баклер Э.С. TASSEL: программное обеспечение для картирования ассоциаций сложных признаков в различных образцах. Биоинформатика. 2007;23(19):2633–5.
КАС Статья пабмед Google ученый
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, et al. TASSEL-GBS: высокопроизводительный конвейер для генотипирования путем секвенирования. ПЛОС Один. 2014;9(2), e
.Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Стеки: набор инструментов для анализа популяционной геномики. Мол Экол. 2013;22(11):3124–40. doi: 10.1111/mec.12354.
Артикул пабмед ПабМед Центральный Google ученый
Лу Ф., Липка А.Е., Глаубиц Дж., Элшир Р., Черней Дж.Х. и др. Геномное разнообразие, плоидность и эволюция проса проса: новые выводы из сетевого протокола обнаружения SNP. Генетика PLoS. 2013;9(1), e1003215. doi: 10.1371/journal.pgen.1003215.
КАС Статья пабмед ПабМед Центральный Google ученый
Гомперт З., Фористер М.Л., Фордайс Дж.А., Найс К.С., Уильямсон Р.Дж., Бюркл К.А. Байесовский анализ молекулярной дисперсии в пиросеквенциях дает количественную оценку генетической структуры популяции в геноме бабочек Lycaeides.
Мол Экол. 2010;19:2455–73.КАС Статья пабмед Google ученый
Линч М. Оценка частот аллелей в проектах секвенирования генома с широким охватом. Генетика. 2009; 182: 295–301.
КАС Статья пабмед ПабМед Центральный Google ученый
Hohenlohe PA, Phillips PC, Cresko WA. Использование популяционной геномики для выявления отбора в природных популяциях: ключевые концепции и методологические соображения. Int J Plant Sci. 2010а;171:1059–1071
Li H, Durbin R. Быстрое и точное выравнивание коротких считываний с преобразованием Берроуза-Уилера. Биоинформатика. 2009; 25:1754–60.
КАС Статья пабмед ПабМед Центральный Google ученый
Сона Х., Бастьен М., Икира Э., Тардивел А., Легаре Г.
и др. Усовершенствованный подход к генотипированию путем секвенирования (GBS), предлагающий повышенную универсальность и эффективность обнаружения SNP и генотипирования. ПЛОС Один. 2013;8(1), e54603.КАС Статья пабмед ПабМед Центральный Google ученый
Torkamaneh D, Belzile F. Сканирование и заполнение: сверхплотное генотипирование SNP, сочетающее генотипирование с помощью секвенирования, массив SNP и данные полногеномного повторного секвенирования. ПЛОС Один. 2015;10(7), e0131533. doi:10.1371/journal.pone.0131533.
Артикул пабмед ПабМед Центральный Google ученый
Felcher KJ, Coombs JJ, Massa AN, Hansey CN, Hamilton JP, et al. Интеграция двух карт сцепления диплоидного картофеля с последовательностью генома картофеля. ПЛОС Один. 2012;7(4), e36347.
КАС Статья пабмед ПабМед Центральный Google ученый
Martin M. Cutadapt удаляет последовательности адаптеров из считываний высокопроизводительного секвенирования. EMBnet J. 2011. doi: http://dx.doi.org/10.14806/ej.17.1.200.
Ли Х. и Дурбин Р. Быстрое и точное выравнивание длинных считываний с помощью преобразования Берроуза-Уилера. Биоинформатика. 2010. Электронная почта. [PMID: 20080505]
Li H. Статистическая основа для определения SNP, обнаружения мутаций, сопоставления ассоциаций и оценки генетических параметров популяции на основе данных секвенирования. Биоинформатика. 2011;27(21):2987–93. PMID: 21
7, Epub 2011, 8 сентября.
CAS Статья пабмед ПабМед Центральный Google ученый
Риммер А., Фан Х., Мэтисон И., Икбал З.
, Твигг С.Р.Ф. и др. Интеграция подходов на основе картирования, сборки и гаплотипов для вызова вариантов в приложениях для клинического секвенирования. Нат Жене. 2014. doi: 10.1038/ng.3036.ПабМед ПабМед Центральный Google ученый
Утконос-Сапело: https://wiki.gacrc.uga.edu/wiki/Утконос-Сапело. По состоянию на 9 декабря 2015 г.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, et al. Формат Variant Call и VCFtools. Биоинформатика. 2011. doi: 10.1093/биоинформатика/btr330.
ПабМед ПабМед Центральный Google ученый
Sonah H, O’Donoughue L, Cober E, Rajcan I, Belzile F. Идентификация локусов, управляющих восемью агрономическими признаками, с использованием подхода GBS-GWAS и проверка картированием QTL в сое. Plant Biotechnol J. 2014. doi: 10.1111/pbi.12249.
ПабМед Google ученый
Mascher M, Wu S, Amand PS, Stein N, Poland J. Применение генотипирования путем секвенирования на полупроводниковых платформах для секвенирования: сравнение генетического и эталонного порядка маркеров в ячмене. ПЛОС Один. 2013;8(10), e76925. doi:10.1371/journal.pone.0076925.
КАС Статья пабмед ПабМед Центральный Google ученый
Peter GV, Uitdewilligen JGAML, Voorrips E, Visser RGF, van Eck HJ. Разработка и анализ массива 20 тыс. SNP для картофеля ( Solanum tuberosum ): знакомство с историей разведения. Теория Appl Genet. 2015. doi:10.1007/s00122-015-2593-y.
Google ученый
Эндельман Дж. Генотипирование путем секвенирования диплоидной популяции картофеля F2. 2015 г. https://pag.confex.com/pag/xxiii/webprogram/Paper15683.html.
Google ученый
Hirsch CN, Hirsch CD, Felcher K, Coombs J, Zarka D, Van Deynze A, De Jong W, Veilleux RE, Jansky S, Bethke P. Ретроспективный взгляд на селекцию североамериканского картофеля (Solanum tuberosum L.) в 20 и 21 веков. 2013. G3 3:1003–1013. дои: 10.1534/G3.113.005595.
Голан Д., Медведев П. Использование конечных автоматов для моделирования процесса секвенирования Ion Torrent и снижения частоты ошибок чтения.
Биоинформатика. 2013;29(13):i344–51. doi: 10.1093/биоинформатика/btt212.КАС Статья пабмед ПабМед Центральный Google ученый
Брэгг Л.М., Стоун Г., Батлер М.К., Хугенхольц П., Тайсон Г.В. Проливая свет на темное секвенирование: описание ошибок в данных Ion Torrent PGM. PLoS Comput Biol. 2013;9(4), e1003031. doi:10.1371/journal.pcbi.1003031.
КАС Статья пабмед ПабМед Центральный Google ученый
Стейси Г. Генетика и геномика сои. Нью-Йорк: Спрингер; 2008. ISBN978-0-387-72299-3.
Чжан Г., Ли С., Лю С. Успехи в науках о ячмене. Дордрехт, Гейдельберг, Нью-Йорк: Springer; 2013. ISBN 978-94-007-4682-4.
Брейдин Дж. М., Коул К. Генетика, геномика и селекция картофеля. Нью-Хэмпшир: Энфилд, CRC Press; 2011. ISBN 9781578087150.
 “>
“>Metzker ML. Технологии секвенирования – следующее поколение. Нат Рев Жене. 2010: 31–46. дои: 10.1038/nrg2626
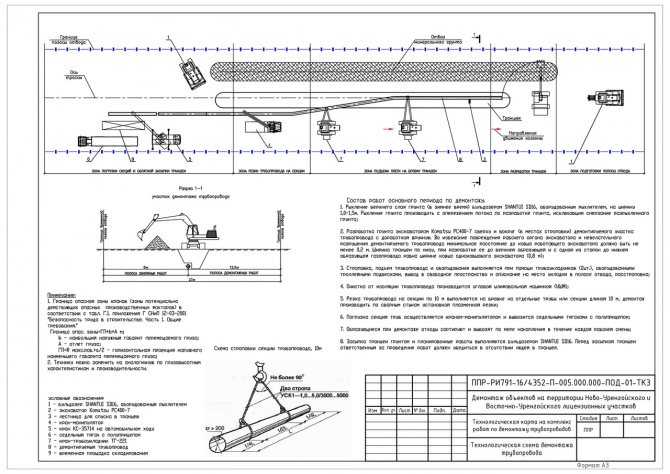 Открытие полногеномных генетических маркеров и генотипирование с использованием секвенирования нового поколения. Природа. 2011. doi: 10.1038/nrg3012.
Открытие полногеномных генетических маркеров и генотипирование с использованием секвенирования нового поколения. Природа. 2011. doi: 10.1038/nrg3012. Надежный и простой подход генотипирования путем секвенирования (GBS) для видов с большим разнообразием. ПЛОС Один. 2011;6(5):19379.
Надежный и простой подход генотипирования путем секвенирования (GBS) для видов с большим разнообразием. ПЛОС Один. 2011;6(5):19379.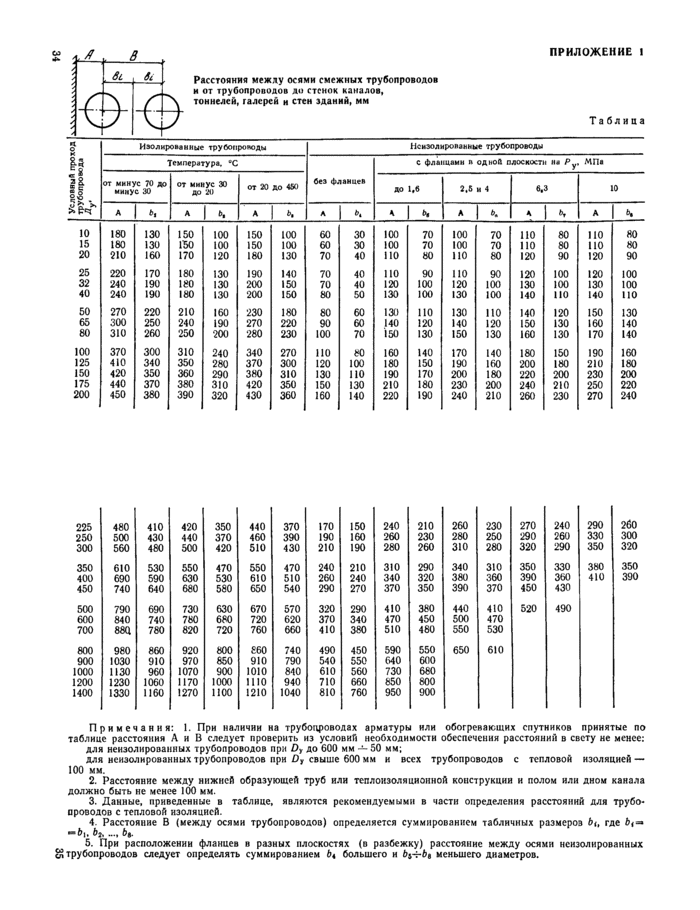 Нат Методы. 2012; 9: 808–10.
Нат Методы. 2012; 9: 808–10. doi:10.1371/journal.pone.00.
doi:10.1371/journal.pone.00.Артикул пабмед ПабМед Центральный Google ученый
 Мол Экол. 2010;19:2455–73.
Мол Экол. 2010;19:2455–73.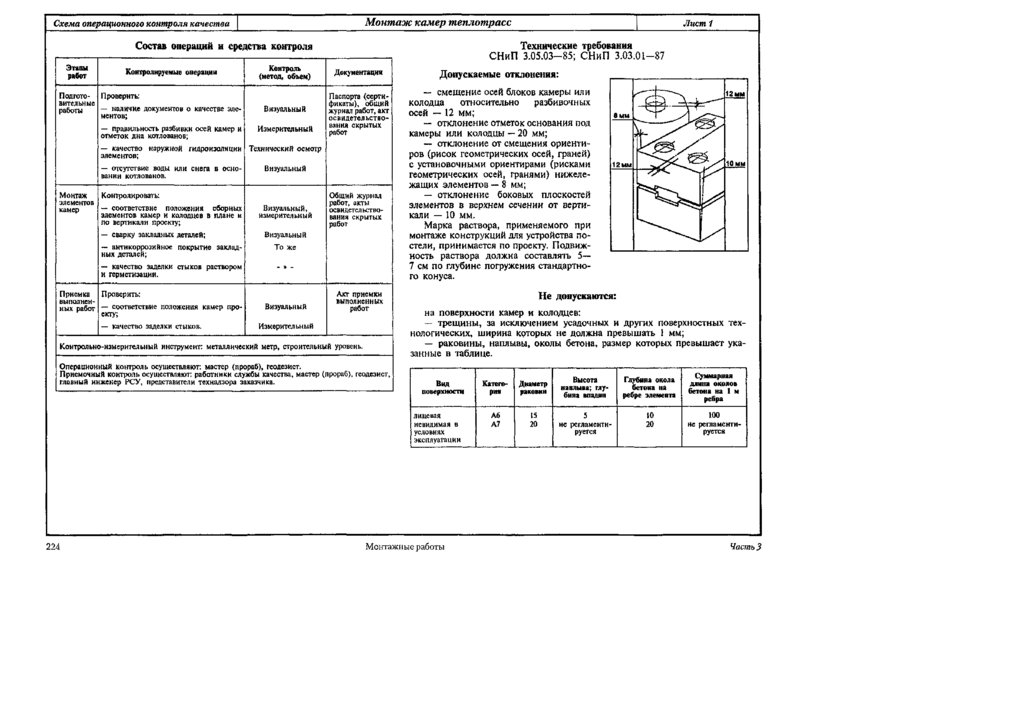 и др. Усовершенствованный подход к генотипированию путем секвенирования (GBS), предлагающий повышенную универсальность и эффективность обнаружения SNP и генотипирования. ПЛОС Один. 2013;8(1), e54603.
и др. Усовершенствованный подход к генотипированию путем секвенирования (GBS), предлагающий повышенную универсальность и эффективность обнаружения SNP и генотипирования. ПЛОС Один. 2013;8(1), e54603.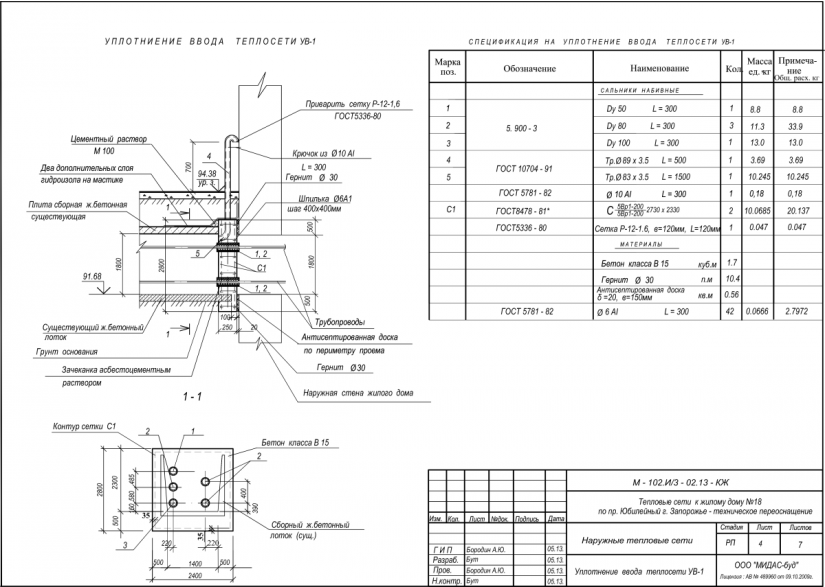 “>
“>Sabre-демультиплексирование штрих-кода: https://github.com/najoshi/sabre. По состоянию на 27 сентября 2013 г.
 , Твигг С.Р.Ф. и др. Интеграция подходов на основе картирования, сборки и гаплотипов для вызова вариантов в приложениях для клинического секвенирования. Нат Жене. 2014. doi: 10.1038/ng.3036.
, Твигг С.Р.Ф. и др. Интеграция подходов на основе картирования, сборки и гаплотипов для вызова вариантов в приложениях для клинического секвенирования. Нат Жене. 2014. doi: 10.1038/ng.3036. “>
“>Международный консорциум по секвенированию генома ячменя. Сборка физической, генетической и функциональной последовательности генома ячменя. Природа. 2012; 491:711–6. дои: 10.1038/природа11543.
Google ученый
 “>
“>Прашар А., Хорник С., Янг В., Маклин К., Шарма С.К., Дейл М.Ф., Брайан Г.Дж. Построение плотной карты SNP высокогетерозиготной диплоидной популяции картофеля и анализ QTL формы клубня и глубины глазка. Теория Appl Genet. 2014;127(10):2159–71. doi: 10.1007/s00122-014-2369-9.
Артикул пабмед Google ученый
 Биоинформатика. 2013;29(13):i344–51. doi: 10.1093/биоинформатика/btt212.
Биоинформатика. 2013;29(13):i344–51. doi: 10.1093/биоинформатика/btt212.
Ссылки на скачивание
Благодарности
Авторы хотели бы отметить превосходное обслуживание, полученное от Plate-forme d’analyses génomiques в Университете Лаваля как для подготовки библиотеки GBS, так и для секвенирования.
Финансирование
Финансирование этого исследования было предоставлено Министерством сельского хозяйства и сельского хозяйства Канады и Канадским альянсом по исследованию полевых культур (грант № AIP-CL23).
Вклад авторов
DT, JL и FB задумали проект. JL и DT внесли свой вклад в программирование. АА оценил образцы ячменя. МБ оценил образцы картофеля. DT и FB участвовали в оценке инструментов и написании рукописи. Все авторы прочитали и одобрили окончательный вариант рукописи.
Конкурирующие интересы
Авторы заявляют об отсутствии конкурирующих интересов.
Согласие на публикацию
Неприменимо.
Одобрение этики и согласие на участие
Неприменимо.
Информация об авторе
Авторы и организации
Факультет физиологии, Университет Лаваль, Квебек, Квебек, Канада
Давуд Торкамане, Максим Бастьен, Амина Абед и Франсуа Белзи
Institut de Biologie Intégrative et des Systèmes (IBIS), Université Laval, Quebec City, QC, Canada
Davoud Torkamaneh, Jérôme Laroche, Maxime Bastien, Amina Abed & François Belzile
Authors
- Davoud Torkamaneh
Посмотреть публикации автора
Вы также можете искать этого автора в PubMed Google Scholar
- Jérôme Laroche
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Академия
- Максим Бастьен
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
- Амина Абед
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
- François Belzile
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
Автор, ответственный за переписку
Франсуа Белзил.
Права и разрешения
Открытый доступ Эта статья распространяется в соответствии с условиями международной лицензии Creative Commons Attribution 4.0 (http://creativecommons.org/licenses/by/4.0/), которая разрешает неограниченное использование, распространение и воспроизведение на любом носителе при условии вы должным образом указываете автора (авторов) и источник, предоставляете ссылку на лицензию Creative Commons и указываете, были ли внесены изменения. Отказ от права Creative Commons на общественное достояние (http://creativecommons.org/publicdomain/zero/1.0/) применяется к данным, представленным в этой статье, если не указано иное.
Перепечатки и разрешения
Об этой статье
Правильные и воспроизводимые результаты — документация CFSAN SNP Pipeline 2.2.1
Наша цель — сделать результаты SNP Pipeline полностью
воспроизводимым и столь же правильным, как современное научное понимание
позволяет. В рамках этих усилий мы документируем здесь проблемы, которые у нас есть. обнаружил, что повлияло на правильность. Мы также подробно описываем, как мы строили
это программное обеспечение, чтобы результаты были максимально воспроизводимыми. Кроме того,
мы создаем, собираем и сопоставляем наборы данных, которые мы используем для
оценить правильность и воспроизводимость нашего программного обеспечения. Как
проект, который стал возможен только благодаря самым последним разработкам в
науки и техники, наши усилия по обеспечению правильности и
воспроизводимость являются постоянными усилиями. Издания, которые у нас есть
созданные с целью обеспечения научной правильности, перечислены как
ссылки внизу этого документа. Этот документ будет продолжен
развиваться по мере того, как мы улучшаем наш процесс и по мере того, как происходят научные достижения.
обнаружил, что повлияло на правильность. Мы также подробно описываем, как мы строили
это программное обеспечение, чтобы результаты были максимально воспроизводимыми. Кроме того,
мы создаем, собираем и сопоставляем наборы данных, которые мы используем для
оценить правильность и воспроизводимость нашего программного обеспечения. Как
проект, который стал возможен только благодаря самым последним разработкам в
науки и техники, наши усилия по обеспечению правильности и
воспроизводимость являются постоянными усилиями. Издания, которые у нас есть
созданные с целью обеспечения научной правильности, перечислены как
ссылки внизу этого документа. Этот документ будет продолжен
развиваться по мере того, как мы улучшаем наш процесс и по мере того, как происходят научные достижения.
Воспроизводимые результаты
Мы сделали результаты SNP Pipeline полностью воспроизводимыми, а не только
итоговая матрица SNP, но и каждый промежуточный файл. Воспроизводимые результаты
помогите нам протестировать и отладить конвейер, а также облегчите совместную работу
между исследователями.
Публичная доступность
Исходный код SNP Pipeline доступен на GitHub, так что каждый может загрузить наш исходный код. Репозиторий GitHub содержит некоторые наборы данных, которые можно использовать для воспроизвести выбранные результаты. Мы также предоставляем информацию о том, как получить и проверить другие наборы данных, которые мы использовали. (Эти наборы данных большие, поэтому мы не предоставляйте их напрямую.)
Контроль версий
Мы используем git для внутренней разработки кода, чтобы обеспечить контроль над наш исходный код и может определить, какая версия кода использовалась для создания любого конкретный результат. Мы помечаем/версионируем коммиты кода, который считаем готовым релизы и использовать их для большинства наших внутренних анализов. Мы также выпускаем каждую из этих помеченных версий на GitHub и в индекс пакетов Python для удобства. монтаж.
Параметры
Поведение конвейера SNP зависит от настройки ряда параметров, которые
определить поведение различных программных пакетов, которые использует конвейер. Эти
параметры влияют как на правильность, так и на воспроизводимость результатов. Мы установили
все параметры, чтобы результаты были воспроизводимыми. Это влечет за собой
установка семян для всех процессов, зависящих от случайных чисел, а также конкретных
выбор других параметров, которые могут повлиять на такое поведение, как порядок
полученные результаты. Мы обсудим эти аспекты обеспечения воспроизводимости более подробно в
другие части этого документа.
Эти
параметры влияют как на правильность, так и на воспроизводимость результатов. Мы установили
все параметры, чтобы результаты были воспроизводимыми. Это влечет за собой
установка семян для всех процессов, зависящих от случайных чисел, а также конкретных
выбор других параметров, которые могут повлиять на такое поведение, как порядок
полученные результаты. Мы обсудим эти аспекты обеспечения воспроизводимости более подробно в
другие части этого документа.
Конвейер зависит от некоторых довольно сложных программных пакетов, и эти пакеты имеют
большое количество параметров. Как выпущено, конвейер не указывает значения
для всех возможных параметров, но только для тех, которые мы сочли полезным изменить в нашей работе.
Файл конфигурации, используемый конвейером, содержит запись используемых параметров.
а также дает возможность настраивать поведение конвейера, добавляя или
изменение параметров по мере необходимости. Те, кто желает дополнительно изменить
поведение программного обеспечения придется корректировать код в соответствии со своими потребностями.
Мы рекомендуем сохранить значения параметров, используемые для любых важных результатов, в идеале в сценарии или файле конфигурации, который находится под контролем версий. Конвейер генерирует лог-файлы, документирующие каждый запуск путем записи версий программного обеспечения и параметров, используемых для каждый запуск.
Параллелизм
Конвейер SNP использует преимущества нескольких ядер ЦП для выполнения частей обработка параллельно. Однако параллелизм может привести к недетерминированному поведению. и разные результаты при повторном запуске конвейера. Адреса конвейера известные проблемы параллелизма с бабочкой и samtools.
Галстук-бабочка
Конвейер SNP использует несколько ядер ЦП во время выравнивания галстука-бабочки. Если не сказали
в противном случае, когда бабочка запускает несколько параллельных потоков, она генерирует выходные записи.
в файле SAM в недетерминированном порядке. Следствием этого является ЗУР
файлы и файлы Pileup могут различаться между запусками. Это может выглядеть как два соседних
базы чтения поменялись местами в файлах пайлапа.
Это может выглядеть как два соседних
базы чтения поменялись местами в файлах пайлапа.
Чтобы обойти эту проблему, конвейер использует командную строку --reorder Bowtie.
вариант. Опция переупорядочивания заставляет бабочку генерировать выходные записи в том же порядке.
как читает во входном файле. Это обсуждается в документации по бабочке здесь:
http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml#performance-options
SAMtools
Конвейер SNP одновременно запускает несколько процессов samtools для создания
пайлапы для каждого семпла. Когда запускается процесс пайлапа samtools, он проверяет
наличие ссылочного файла faidx, *.fai . Если файл faidx не существует,
samtools создает его автоматически. Однако несколько процессов samtools mpileup могут
мешают друг другу, пытаясь одновременно создать файл. Этот
помехи вызывают неправильные результаты конвейера.
Чтобы обойти эту проблему, конвейер явно создает файл faidx
файл, запустив samtools faidx по ссылке перед запуском
процессы компиляции. Это предотвращает ошибки позже, когда несколько samtools
процессы mpileup выполняются одновременно.
Это предотвращает ошибки позже, когда несколько samtools
процессы mpileup выполняются одновременно.
Версии программного обеспечения
Различные версии пакетов программного обеспечения, которые использует этот конвейер, могут генерировать разные результаты. Это важно знать, если вы в конечном итоге сравните результаты между запусками. Делимся своими наблюдениями из версий SAMtools и Bowtie, которые мы использовали ниже.
Bowtie
Различные версии Bowtie могут генерировать разные файлы SAM, которые впоследствии вызывает различные наложения и обнаружение различных вариантов. Например, с нашими включенными наборами данных Bowtie 2.1.0 и 2.2.2 производят функционально идентичные файлы SAM при работе с набором данных Lambda Virus. Однако сгенерированные файлы SAM (и последующие результаты) отличаются при работе с набором данных Salmonella Agona.
SAMtools
SAMtools mpileup версии 0.1.18 и версии 0.1.19различаются по своим
поведение по умолчанию. Версия 0.1.19 умеет отфильтровывать базы с низким
качества, и по умолчанию исключаются базы с показателем качества ниже 13.
(точность 95%). В версии 0.1.18 такой возможности нет, поэтому
разные версии SAMtools mpileup при запуске с настройками по умолчанию
параметры могут создавать различные файлы наложения, которые могут повлиять на snp
список и матрица snp.
Версия 0.1.19 умеет отфильтровывать базы с низким
качества, и по умолчанию исключаются базы с показателем качества ниже 13.
(точность 95%). В версии 0.1.18 такой возможности нет, поэтому
разные версии SAMtools mpileup при запуске с настройками по умолчанию
параметры могут создавать различные файлы наложения, которые могут повлиять на snp
список и матрица snp.
В одном из наших наборов данных из 116 образцов мы наблюдали следующие результаты:
- 36030 snps найдено при создании пайлапов с помощью SAMtools 0.1.18
- 38154 snps найдено при создании пайлапов с помощью SAMtools 0.1.19
Наборы тестовых данных
Мы создали несколько наборов данных для использования при тестировании как воспроизводимость и корректность конвейера. В следующих разделах мы кратко опишем эти наборы данных.
Лямбда-вирус
Этот набор данных был создан с использованием примера Bowtie2 и предназначался для небольшого
тестовый пример и пример, который быстро запустится и проверит базовую функциональность
кода.